

Lipopolysaccharide-Induced Increase in Intestinal Permeability Is Mediated by TAK-1 Activation of IKK and MLCK/MYLK Gene
- PMID: 30711488
- PMCID: PMC6446229
- DOI: 10.1016/j.ajpath.2018.12.016
Lipopolysaccharide-Induced Increase in Intestinal Permeability Is Mediated by TAK-1 Activation of IKK and MLCK/MYLK Gene
Abstract
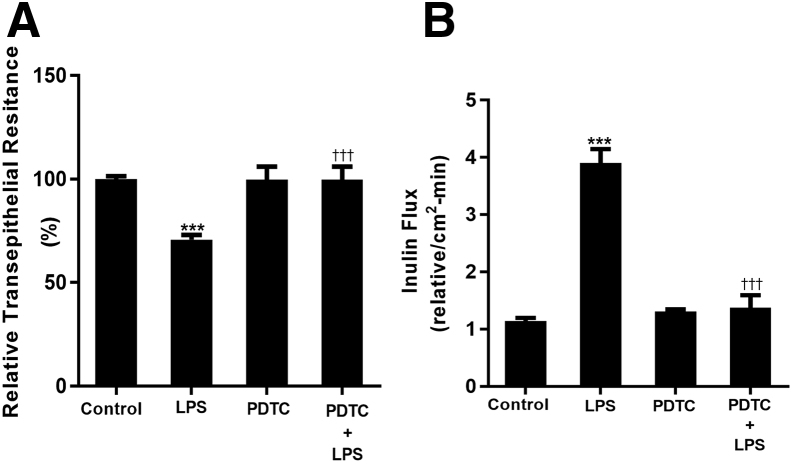
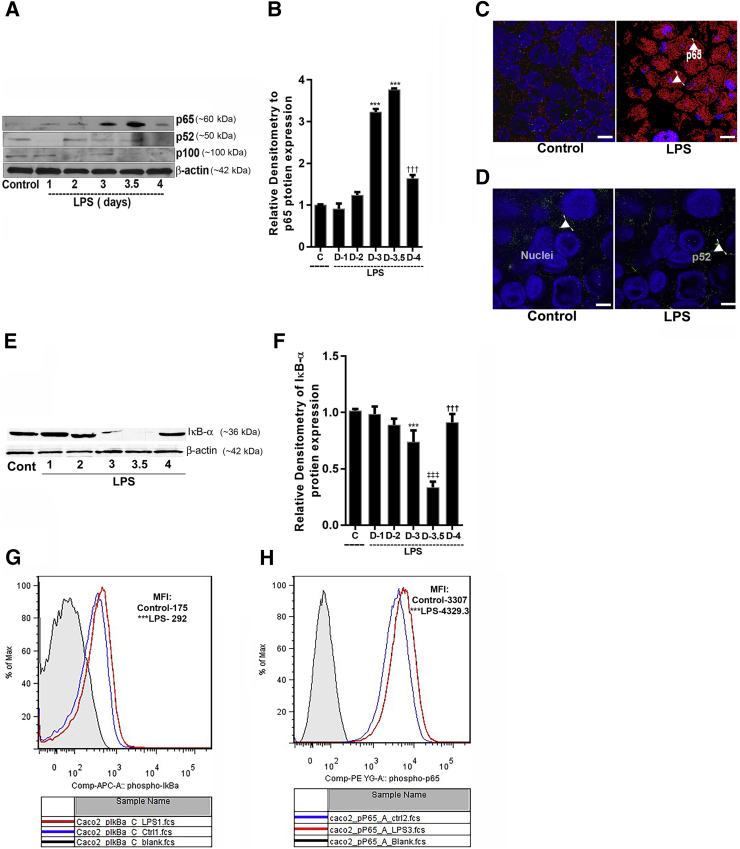
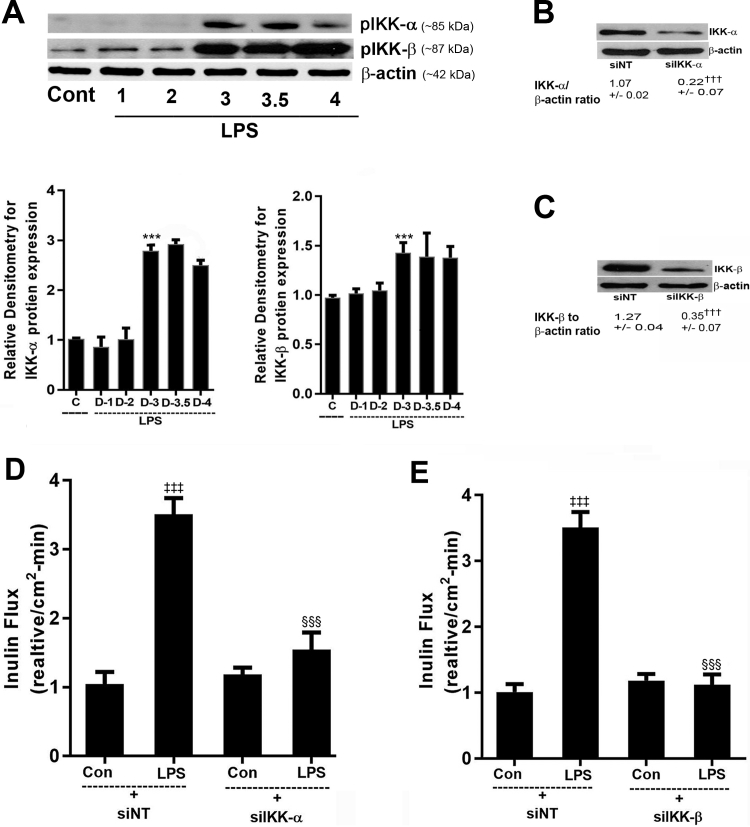
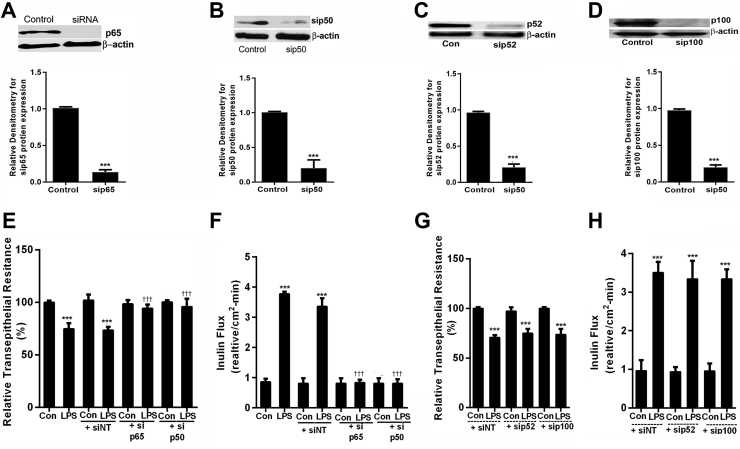
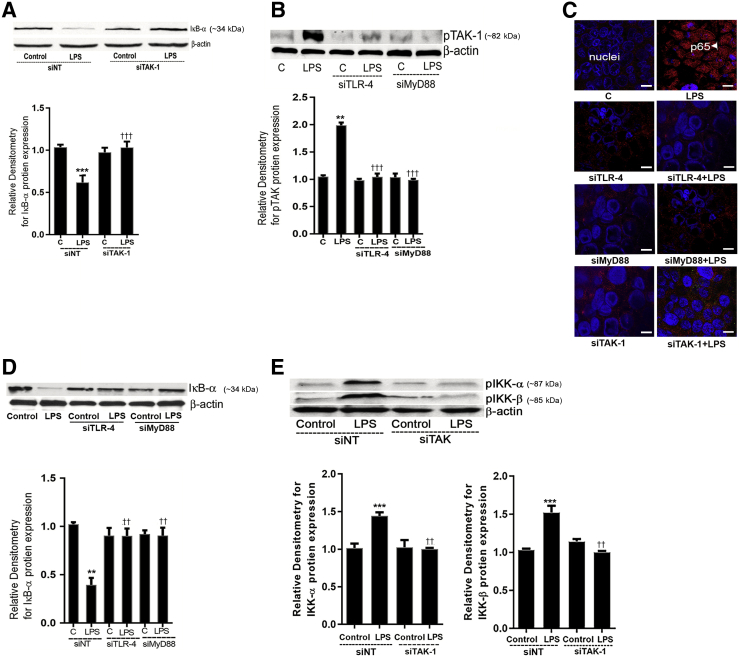
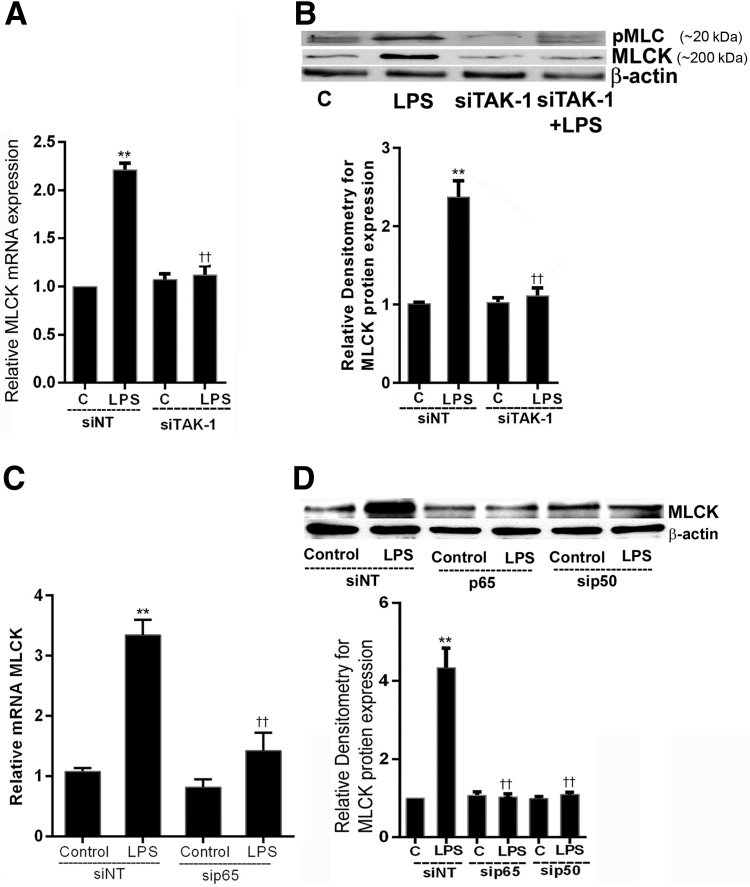
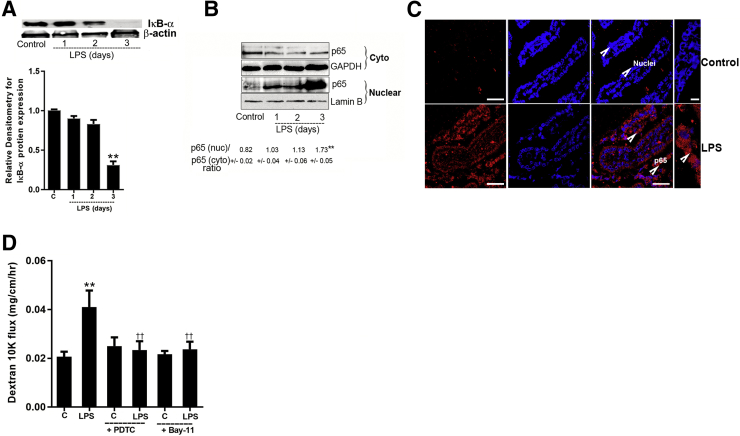
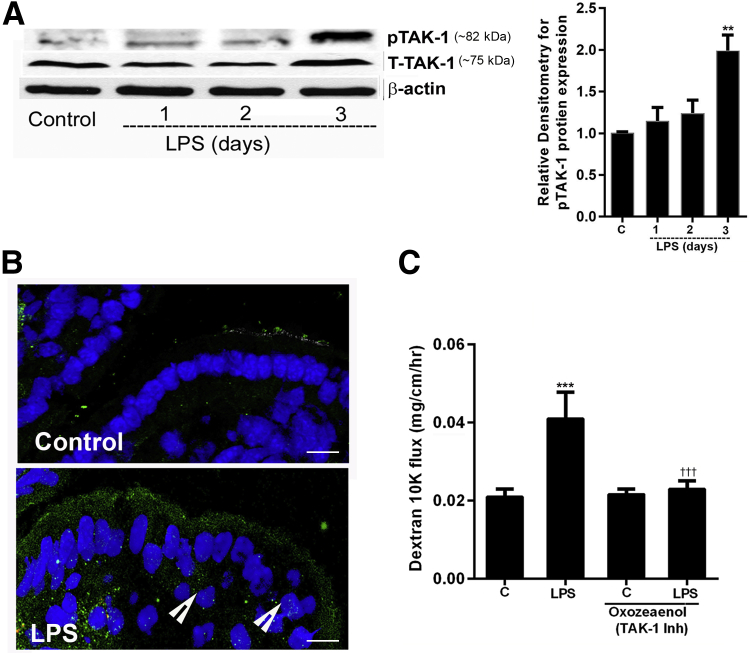
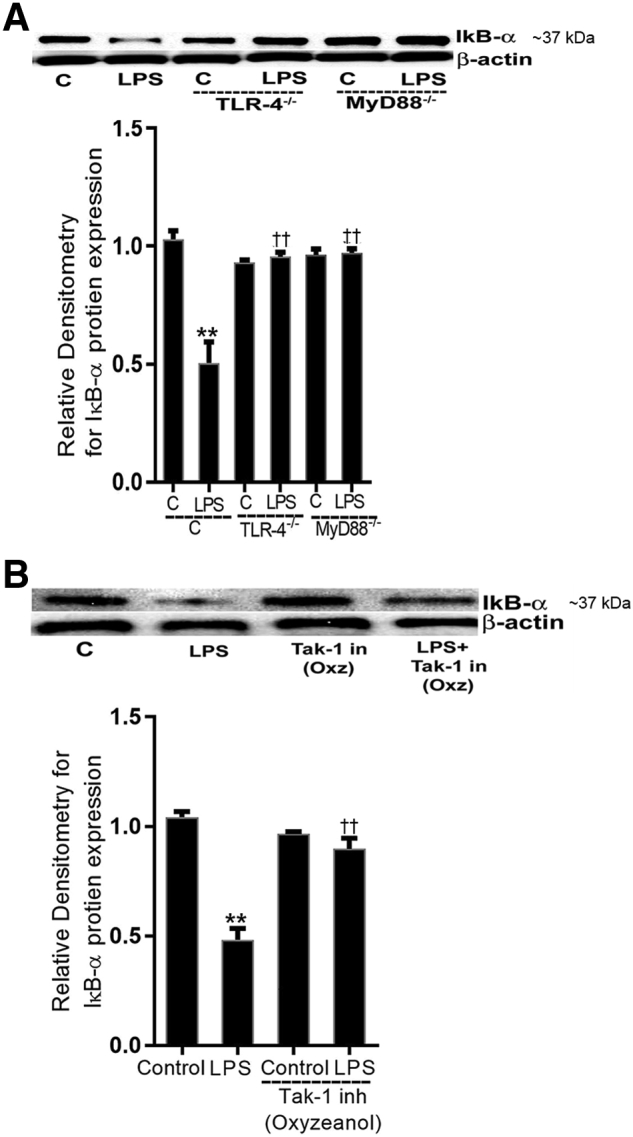
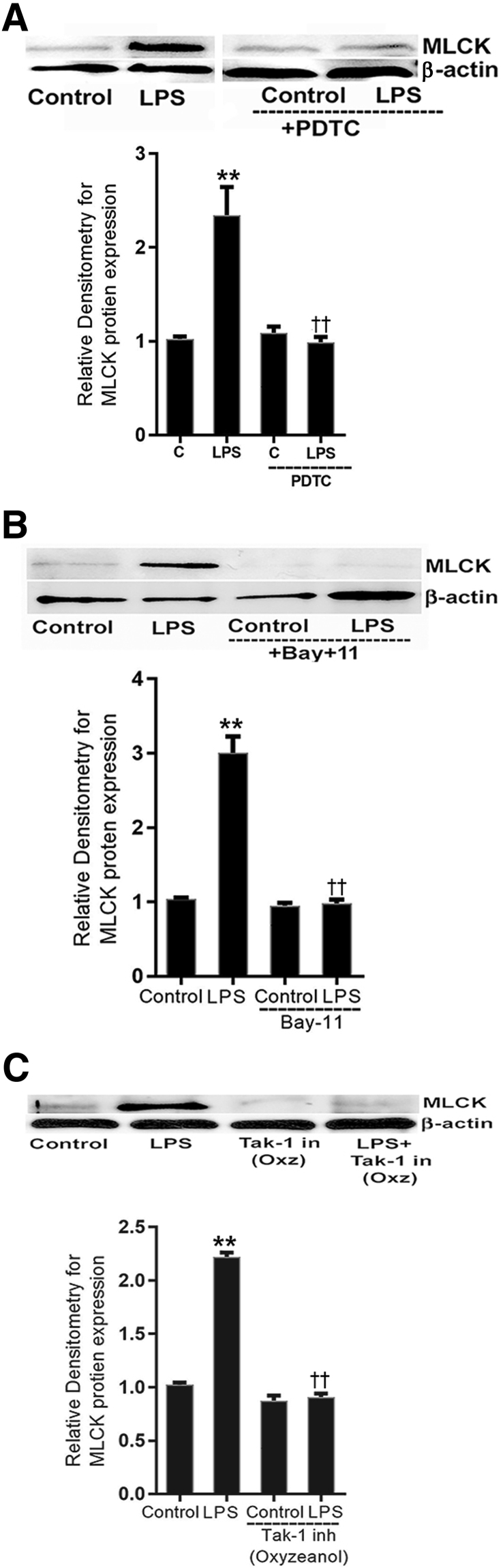
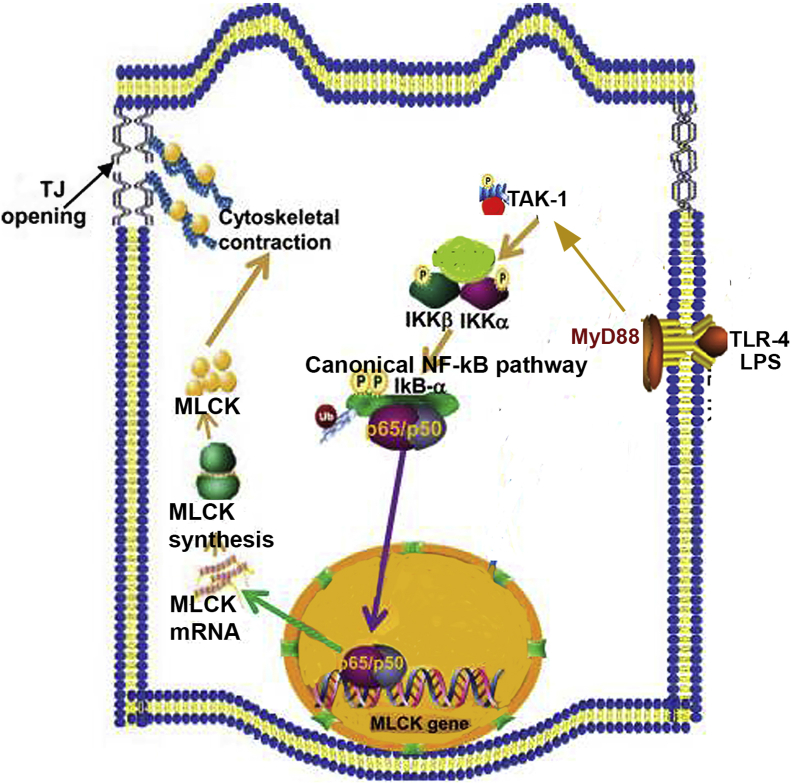
Lipopolysaccharides (LPSs) are a major component of Gram-negative bacterial cell wall and play an important role in promoting intestinal inflammatory responses. Recent studies have shown that physiologically relevant concentrations of LPS (0 to 2000 pg/mL) cause an increase in intestinal epithelial tight junction (TJ) permeability without causing cell death. However, the intracellular pathways and the mechanisms that mediate LPS-induced increase in intestinal TJ permeability remain unclear. The aim was to delineate the intracellular pathways that mediate the LPS-induced increase in intestinal permeability using in vitro and in vivo intestinal epithelial models. LPS-induced increase in intestinal epithelial TJ permeability was preceded by an activation of transforming growth factor-β-activating kinase-1 (TAK-1) and canonical NF-κB (p50/p65) pathways. The siRNA silencing of TAK-1 inhibited the activation of NF-κB p50/p65. The siRNA silencing of TAK-1 and p65/p50 subunit inhibited the LPS-induced increase in intestinal TJ permeability and the increase in myosin light chain kinase (MLCK) expression, confirming the regulatory role of TAK-1 and NF-κB p65/p50 in up-regulating MLCK expression and the subsequent increase in TJ permeability. The data also showed that toll-like receptor (TLR)-4/myeloid differentiation primary response (MyD)88 pathway was crucial upstream regulator of TAK-1 and NF-κB p50/p65 activation. In conclusion, activation of TAK-1 by the TLR-4/MyD88 signal transduction pathway and MLCK by NF-κB p65/p50 regulates the LPS-induced increase in intestinal epithelial TJ permeability.
Copyright © 2019 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures

References
-
- Turner J.R. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. - PubMed
-
- Clark J.A., Doelle S.M., Halpern M.D., Saunders T.A., Holubec H., Dvorak K., Boitano S.A., Dvorak B. Intestinal barrier failure during experimental necrotizing enterocolitis: protective effect of EGF treatment. Am J Physiol Gastrointest Liver Physiol. 2006;291:G938–G949. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
