

MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1
- PMID: 30711629
- PMCID: PMC6904924
- DOI: 10.1053/j.gastro.2019.01.252
MET Inhibitors Promote Liver Tumor Evasion of the Immune Response by Stabilizing PDL1
Abstract
Background & aims: Inhibitors of MET have not produced satisfactory outcomes in trials of patients with liver cancer. We investigated the mechanisms of liver tumor resistance to MET inhibitors in mice.
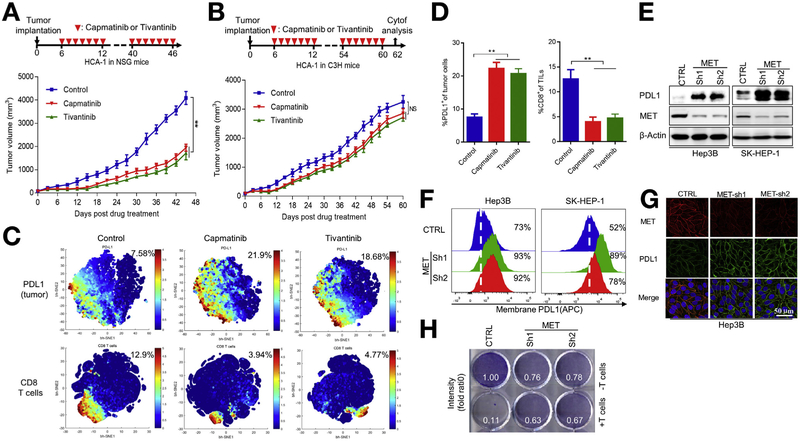
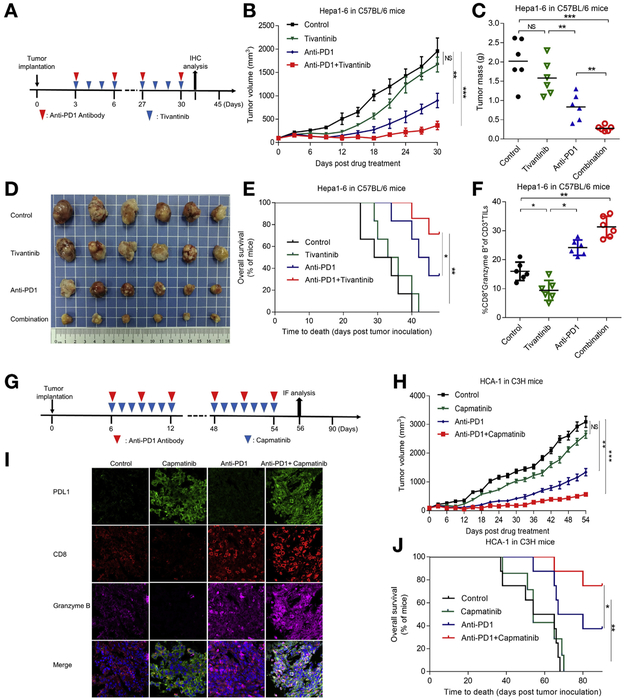
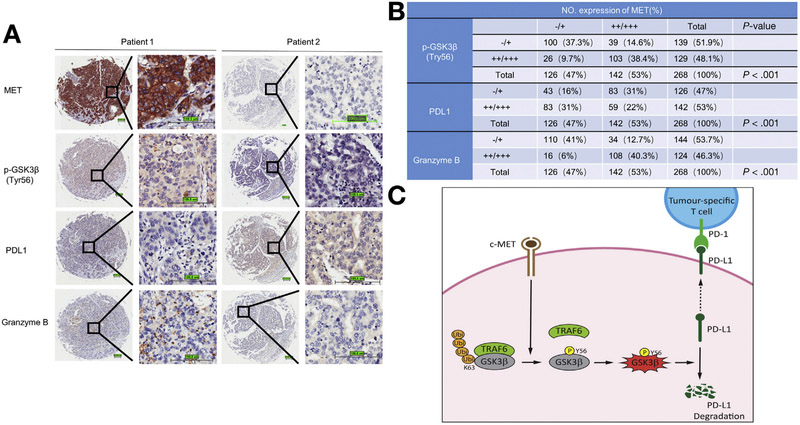
Methods: We tested the effects of MET inhibitors tivantinib and capmatinib in the mouse hepatocellular carcinoma (HCC) cell line HCA-1 and in immune-competent and immunodeficient mice with subcutaneous tumors grown from this cell line. Tumors were collected from mice and tumor cells were analyzed by time-of-flight mass cytometry. We used short hairpin RNAs to weaken expression of MET in Hep3B, SK-HEP-1, HA59T, and HA22T liver cancer cell lines and analyzed cells by immunoblot, immunofluorescence, and immunoprecipitation assays. Mass spectrometry was used to assess interactions between MET and glycogen synthase kinase 3β (GSK3B), and GSK3B phosphorylation, in liver cancer cell lines. C57/BL6 mice with orthotopic tumors grown from Hep1-6 cells were given combinations of capmatinib or tivantinib and antibodies against programmed cell death 1 (PDCD1; also called PD1); tumors were collected and analyzed by immunofluorescence. We analyzed 268 HCCsamples in a tissue microarray by immunohistochemistry.
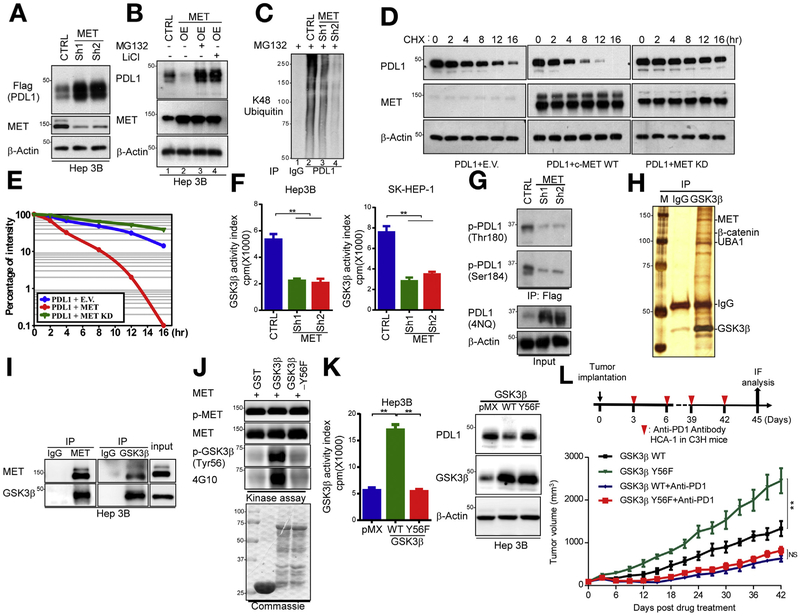
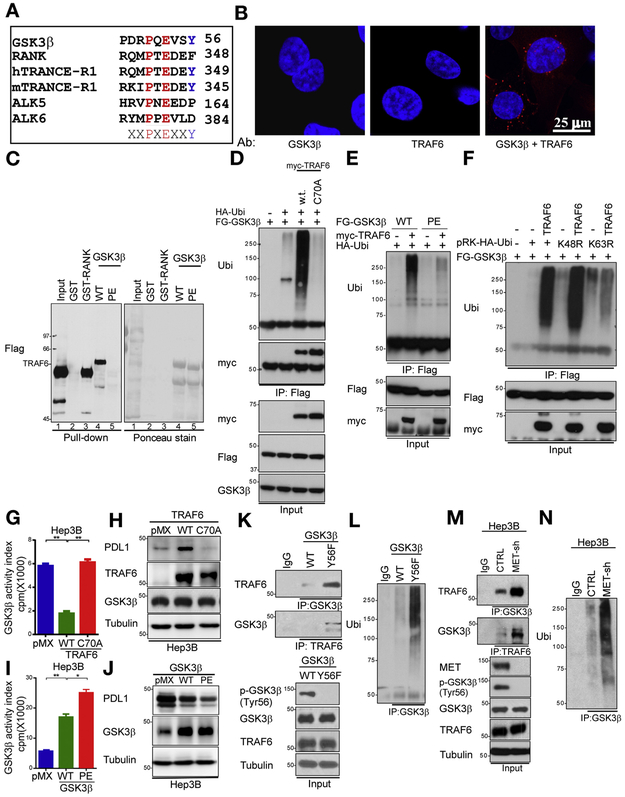
Results: Exposure of liver cancer cell lines to MET inhibitors increased their expression of PD ligand 1 (PDL1) and inactivated cocultured T cells. MET phosphorylated and activated GSK3B at tyrosine 56, which decreased the expression of PDL1 by liver cancer cells. In orthotopic tumors grown in immune-competent mice, MET inhibitors decreased the antitumor activity of T cells. However, addition of anti-PD1 decreased orthotopic tumor growth and prolonged survival of mice compared with anti-PD1 or MET inhibitors alone. Tissue microarray analysis of HCC samples showed an inverse correlation between levels of MET and PDL1 and a positive correlation between levels of MET and phosphorylated GSK3B.
Conclusions: In studies of liver cancer cell lines and mice with orthotopic tumors, MET mediated phosphorylation and activated GSK3B, leading to decreased expression of PDL1. Combined with a MET inhibitor, anti-PD1 and anti-PDL1 produced additive effect to slow growth of HCCs in mice.
Keywords: Glycogen Synthase Kinase 3; Hepatocellular Carcinoma; Programmed Cell Death Ligand 1; Tumor Necrosis Factor Receptor-Associated Factor 6.
Copyright © 2019 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflicts of interest
The authors disclose no competing financial interests (financial, professional, and personal).
Figures
Comment in
-
Has PD-1 MET Its Match in Hepatocellular Carcinoma?Gastroenterology. 2019 May;156(6):1563-1565. doi: 10.1053/j.gastro.2019.03.029. Epub 2019 Mar 26. Gastroenterology. 2019. PMID: 30926349 No abstract available.
References
-
- El-Serag HB. Hepatocellular carcinoma. N Engl J Med 2011;365:1118–1127. - PubMed
-
- Llovet JM, Ricci S, Mazzaferro V, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008; 359:378–390. - PubMed
-
- Bruix J, Qin S, Merle P, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet 2017; 389:56–66. - PubMed
-
- Kudo M, Finn RS, Qin S, et al. Lenvatinib versus sorafenib in first-line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non-inferiority trial. Lancet 2018; 391:1163–1173. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
