

Selective Disruption of Mitochondrial Thiol Redox State in Cells and In Vivo
- PMID: 30713096
- PMCID: PMC6436940
- DOI: 10.1016/j.chembiol.2018.12.002
Selective Disruption of Mitochondrial Thiol Redox State in Cells and In Vivo
Abstract
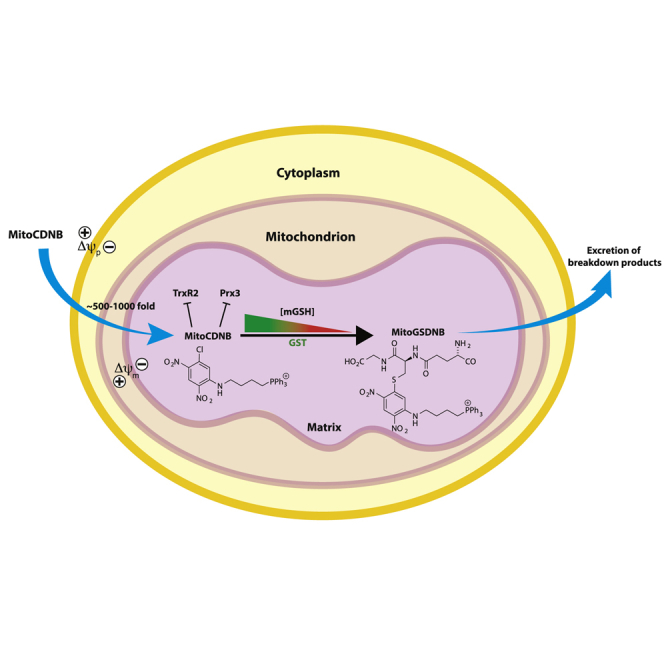
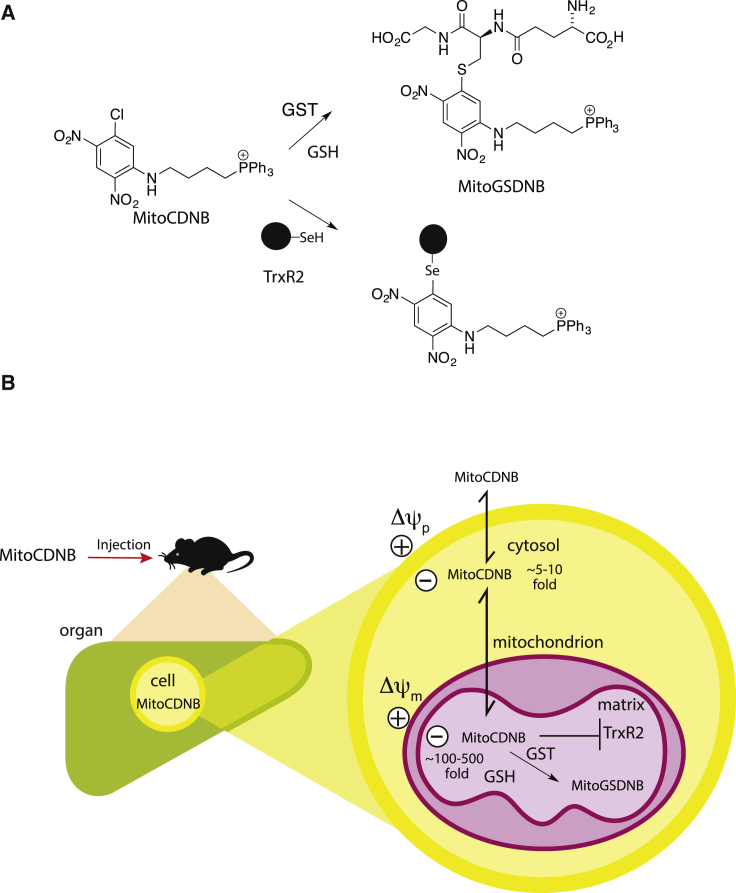
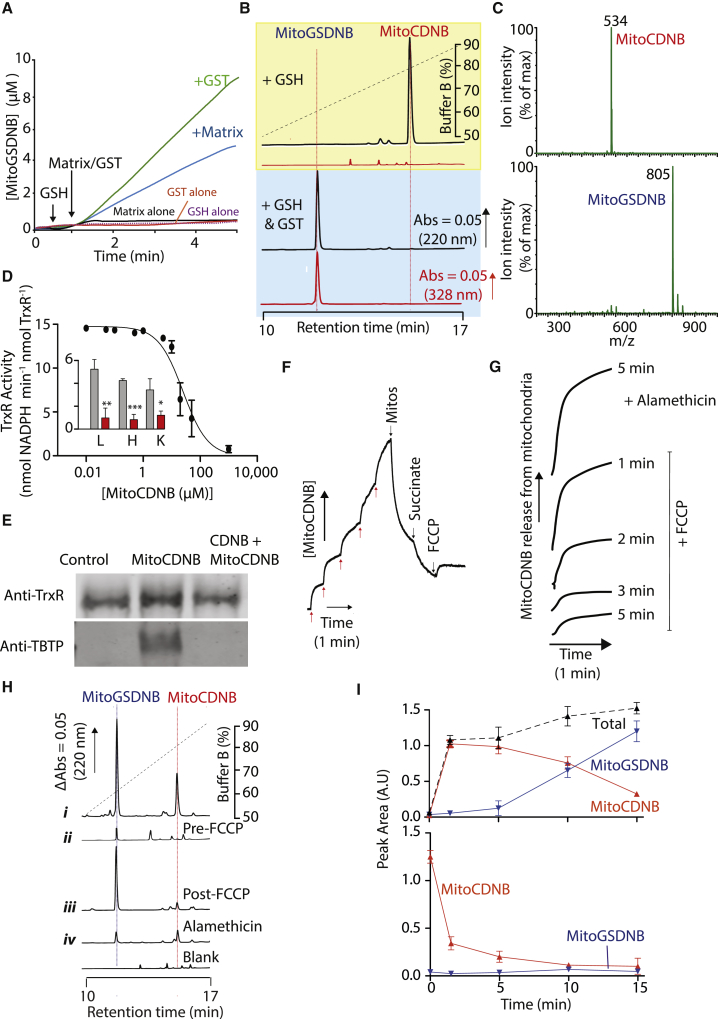
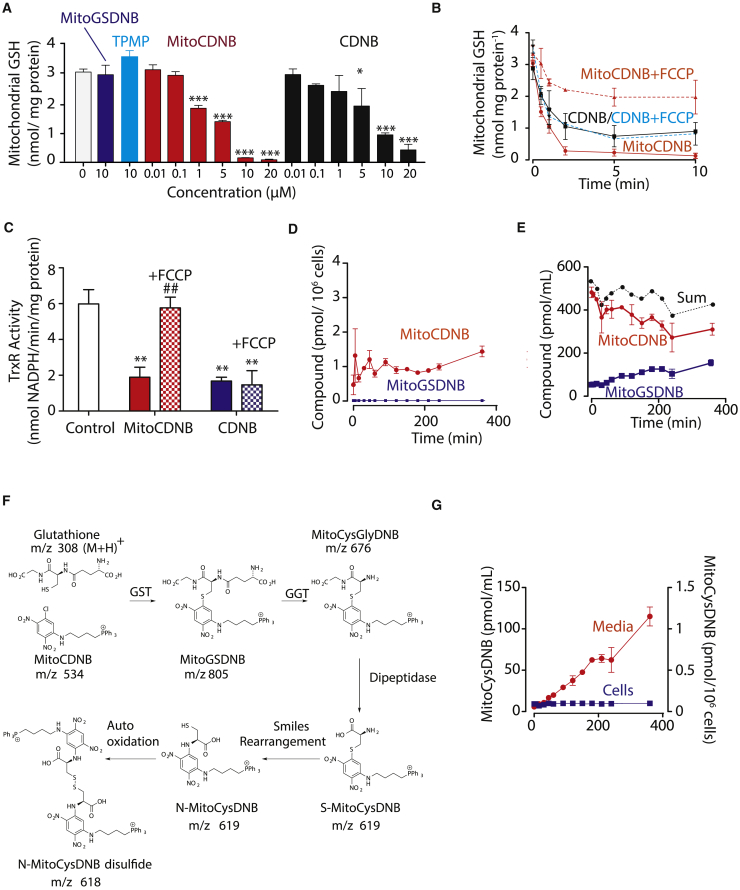
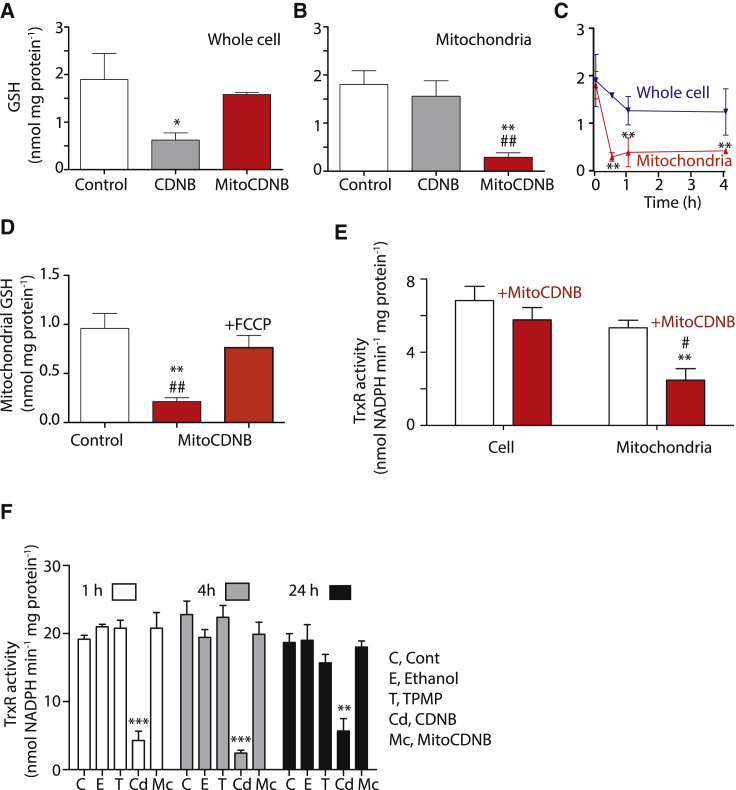
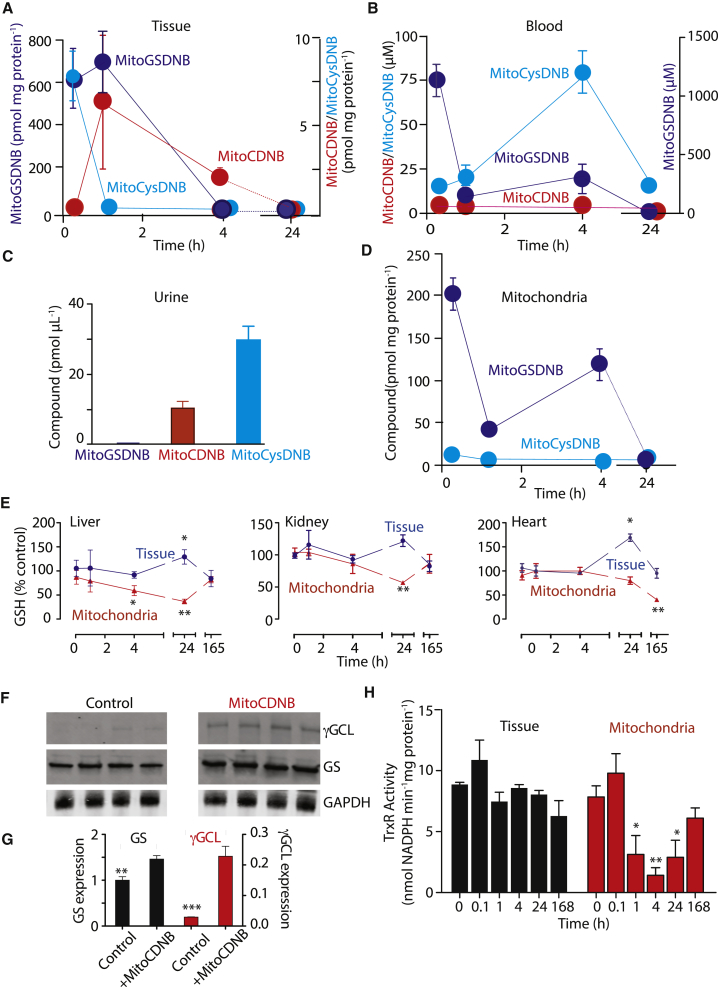
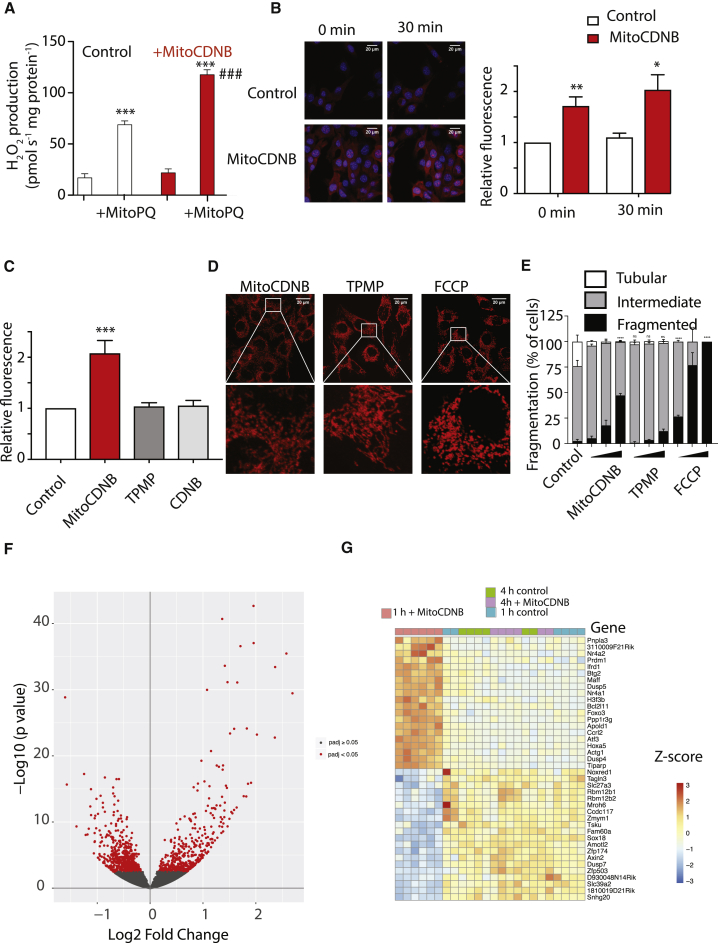
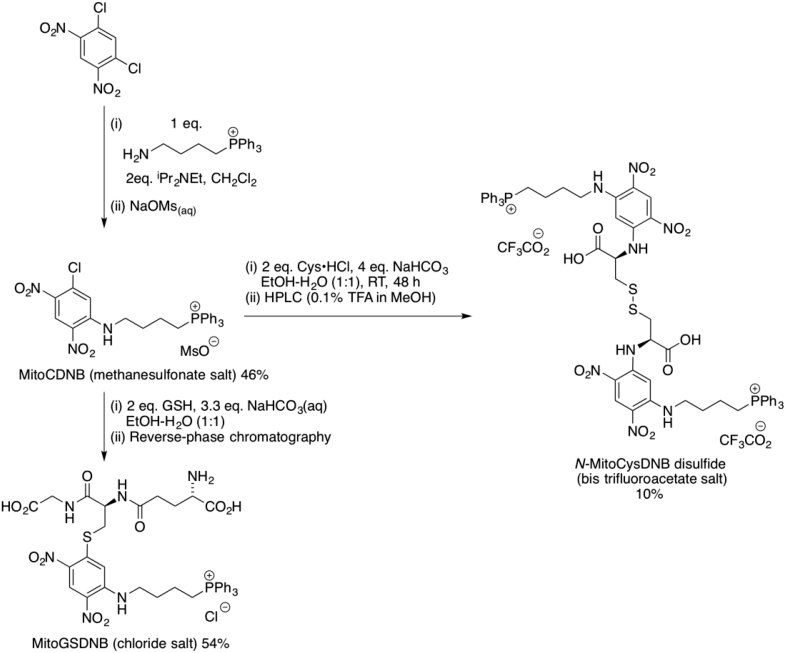
Mitochondrial glutathione (GSH) and thioredoxin (Trx) systems function independently of the rest of the cell. While maintenance of mitochondrial thiol redox state is thought vital for cell survival, this was not testable due to the difficulty of manipulating the organelle's thiol systems independently of those in other cell compartments. To overcome this constraint we modified the glutathione S-transferase substrate and Trx reductase (TrxR) inhibitor, 1-chloro-2,4-dinitrobenzene (CDNB) by conjugation to the mitochondria-targeting triphenylphosphonium cation. The result, MitoCDNB, is taken up by mitochondria where it selectively depletes the mitochondrial GSH pool, catalyzed by glutathione S-transferases, and directly inhibits mitochondrial TrxR2 and peroxiredoxin 3, a peroxidase. Importantly, MitoCDNB inactivates mitochondrial thiol redox homeostasis in isolated cells and in vivo, without affecting that of the cytosol. Consequently, MitoCDNB enables assessment of the biomedical importance of mitochondrial thiol homeostasis in reactive oxygen species production, organelle dynamics, redox signaling, and cell death in cells and in vivo.
Keywords: glutathione; mitochondria; mitochondria targeting; redox signaling; thiol redox state; thioredoxin.
Copyright © 2018 The Author(s). Published by Elsevier Ltd.. All rights reserved.
Figures
Comment in
-
Selectively Addressing Mitochondrial Glutathione and Thioredoxin Redox Systems.Cell Chem Biol. 2019 Mar 21;26(3):316-318. doi: 10.1016/j.chembiol.2019.02.017. Cell Chem Biol. 2019. PMID: 30901559
Similar articles
-
Spatio-temporal changes in glutathione and thioredoxin redox couples during ionizing radiation-induced oxidative stress regulate tumor radio-resistance.Free Radic Res. 2015 Oct;49(10):1218-32. doi: 10.3109/10715762.2015.1056180. Free Radic Res. 2015. PMID: 26021764
-
Nrf2 is activated by disruption of mitochondrial thiol homeostasis but not by enhanced mitochondrial superoxide production.J Biol Chem. 2021 Jan-Jun;296:100169. doi: 10.1074/jbc.RA120.016551. Epub 2020 Dec 13. J Biol Chem. 2021. PMID: 33298526 Free PMC article.
-
Alcohol induces mitochondrial redox imbalance in alveolar macrophages.Free Radic Biol Med. 2013 Dec;65:1427-1434. doi: 10.1016/j.freeradbiomed.2013.10.010. Epub 2013 Oct 16. Free Radic Biol Med. 2013. PMID: 24140864 Free PMC article.
-
Thiol redox control via thioredoxin and glutaredoxin systems.Biochem Soc Trans. 2005 Dec;33(Pt 6):1375-7. doi: 10.1042/BST0331375. Biochem Soc Trans. 2005. PMID: 16246122 Review.
-
Mitochondrial thiols in antioxidant protection and redox signaling: distinct roles for glutathionylation and other thiol modifications.Antioxid Redox Signal. 2012 Mar 15;16(6):476-95. doi: 10.1089/ars.2011.4289. Epub 2011 Dec 20. Antioxid Redox Signal. 2012. PMID: 21954972 Review.
Cited by
-
Mitochondrial outer membrane protein FUNDC2 promotes ferroptosis and contributes to doxorubicin-induced cardiomyopathy.Proc Natl Acad Sci U S A. 2022 Sep 6;119(36):e2117396119. doi: 10.1073/pnas.2117396119. Epub 2022 Aug 29. Proc Natl Acad Sci U S A. 2022. PMID: 36037337 Free PMC article.
-
Insights on Targeting Small Molecules to the Mitochondrial Matrix and the Preparation of MitoB and MitoP as Exomarkers of Mitochondrial Hydrogen Peroxide.Methods Mol Biol. 2021;2275:87-117. doi: 10.1007/978-1-0716-1262-0_6. Methods Mol Biol. 2021. PMID: 34118033
-
Thioredoxin-1 Rescues MPP+/MPTP-Induced Ferroptosis by Increasing Glutathione Peroxidase 4.Mol Neurobiol. 2021 Jul;58(7):3187-3197. doi: 10.1007/s12035-021-02320-1. Epub 2021 Feb 26. Mol Neurobiol. 2021. PMID: 33634378
-
Respiratory chain signalling is essential for adaptive remodelling following cardiac ischaemia.J Cell Mol Med. 2020 Mar;24(6):3534-3548. doi: 10.1111/jcmm.15043. Epub 2020 Feb 10. J Cell Mol Med. 2020. PMID: 32040259 Free PMC article.
-
Unique assembly of carbonylpyridinium and chromene reveals mitochondrial thiol starvation under ferroptosis and novel ferroptosis inducer.Chem Sci. 2022 Mar 2;13(13):3706-3712. doi: 10.1039/d2sc00328g. eCollection 2022 Mar 30. Chem Sci. 2022. PMID: 35432896 Free PMC article.
References
-
- Akerboom T.P.M., Sies H. Assay of glutathione, glutathione disulfide and glutathione mixed disulfides in biological samples. Methods Enzymol. 1981;113:373–382. - PubMed
-
- Arner E.S., Holmgren A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000;267:6102–6109. - PubMed
-
- Arnér E.S.J., Björnstedt M., Holmgren A. 1-Chloro-2,4-dinitrobenzene (DNCB) is an irreversible inhibitor of human thioredoxin reductase: loss of thioredoxin disulfide reductase activity is accompanied by a large increase in NADPH oxidase activity. J. Biol. Chem. 1995;270:3479–3482. - PubMed
-
- Asin-Cayuela J., Manas A.R., James A.M., Smith R.A., Murphy M.P. Fine-tuning the hydrophobicity of a mitochondria-targeted antioxidant. FEBS Lett. 2004;571:9–16. - PubMed
-
- Baars A.J., Mukhtar H., Zoetemelk C.E., Jansen M., Breimer D.D. Glutathione S-transferase activity in rat and human tissues and organs. Comp. Biochem. Physiol. C. 1981;70:285–288. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- MC_ UU_00015/7/MRC_/Medical Research Council/United Kingdom
- MC_UU_00015/3/MRC_/Medical Research Council/United Kingdom
- BB/I012826/1/BB_/Biotechnology and Biological Sciences Research Council/United Kingdom
- MC_UU_00015/7/MRC_/Medical Research Council/United Kingdom
- RG88195/WT_/Wellcome Trust/United Kingdom
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
