

The biosynthetic implications of late-stage condensation domain selectivity during glycopeptide antibiotic biosynthesis
- PMID: 30713624
- PMCID: PMC6333238
- DOI: 10.1039/c8sc03530j
The biosynthetic implications of late-stage condensation domain selectivity during glycopeptide antibiotic biosynthesis
Abstract
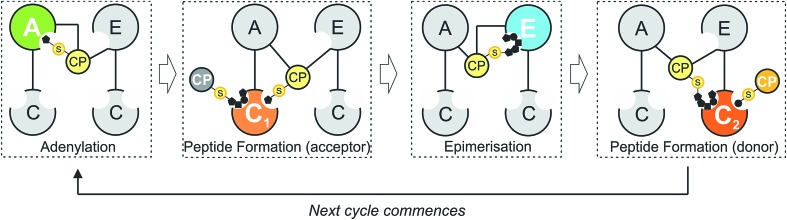
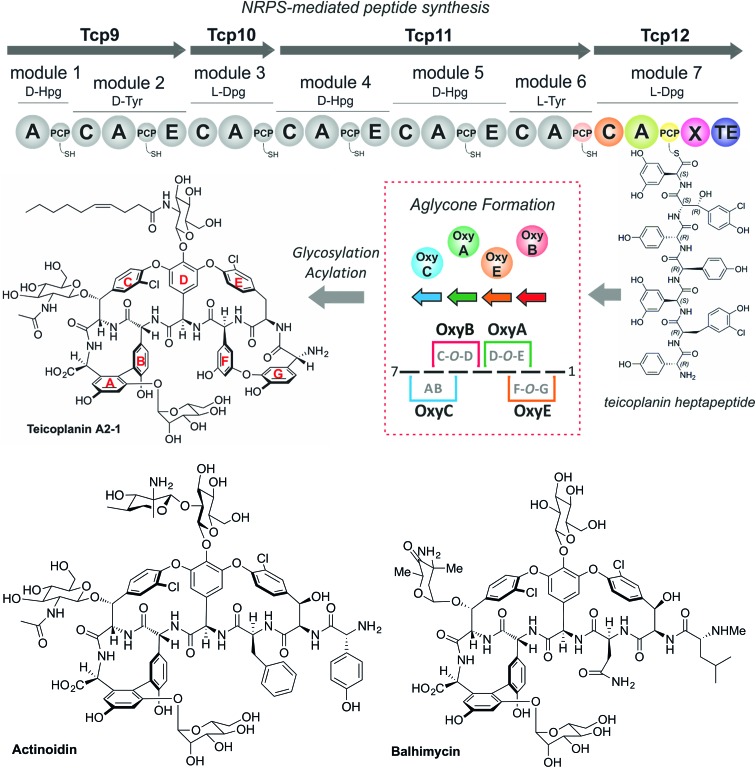
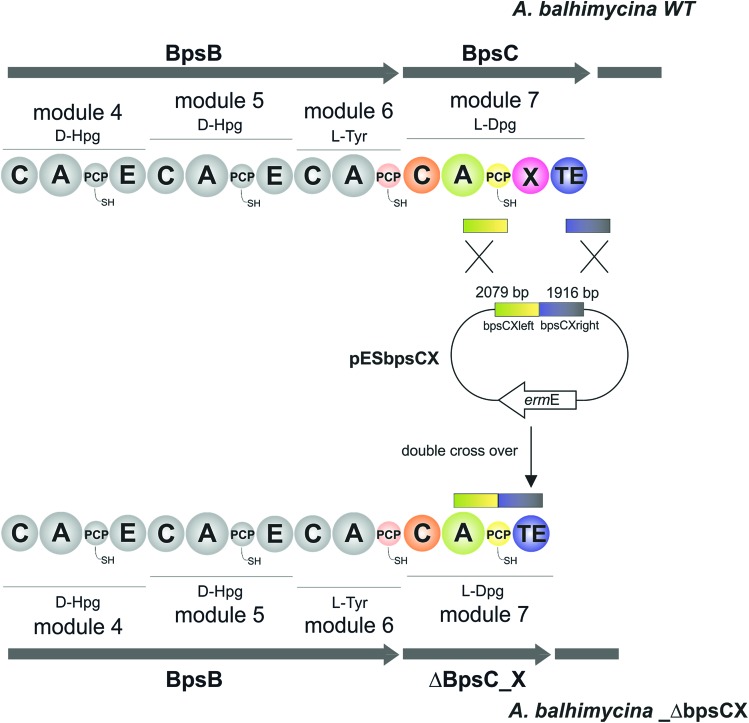
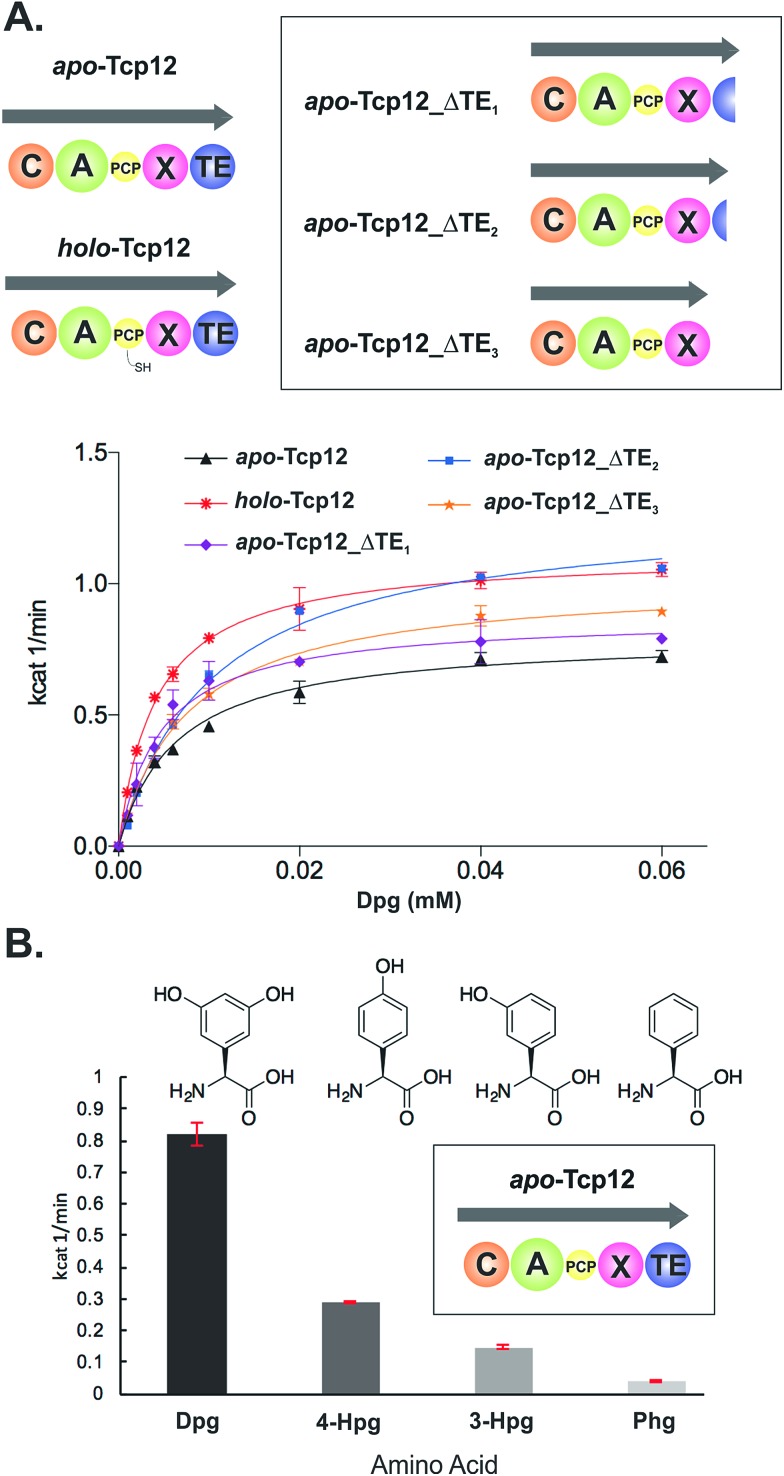
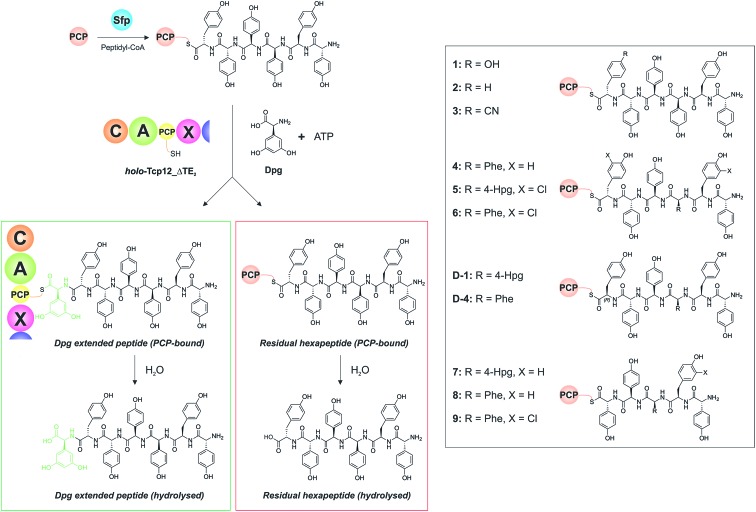
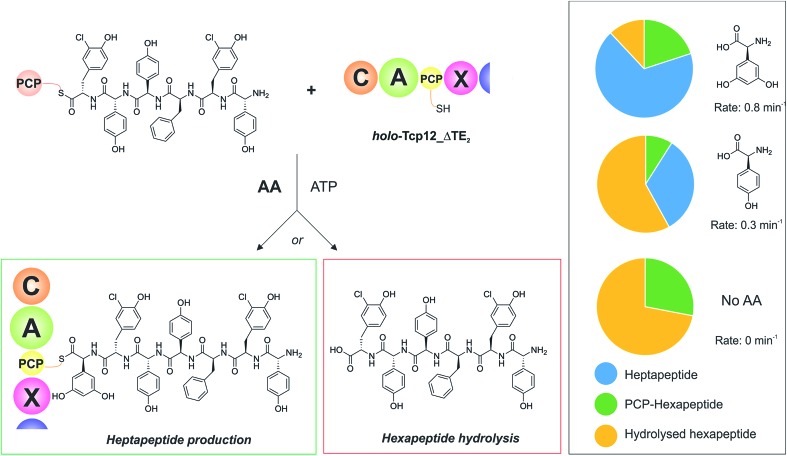
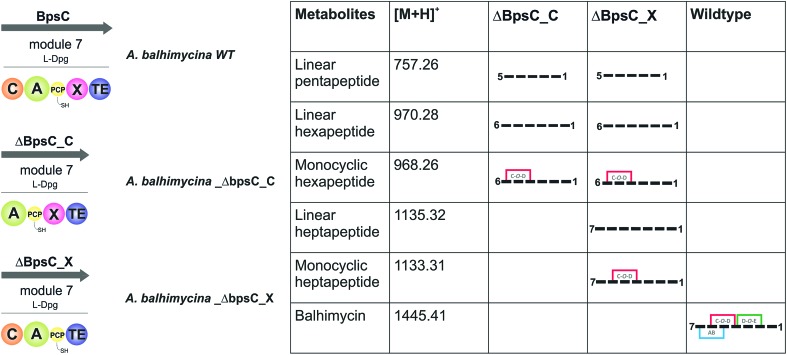
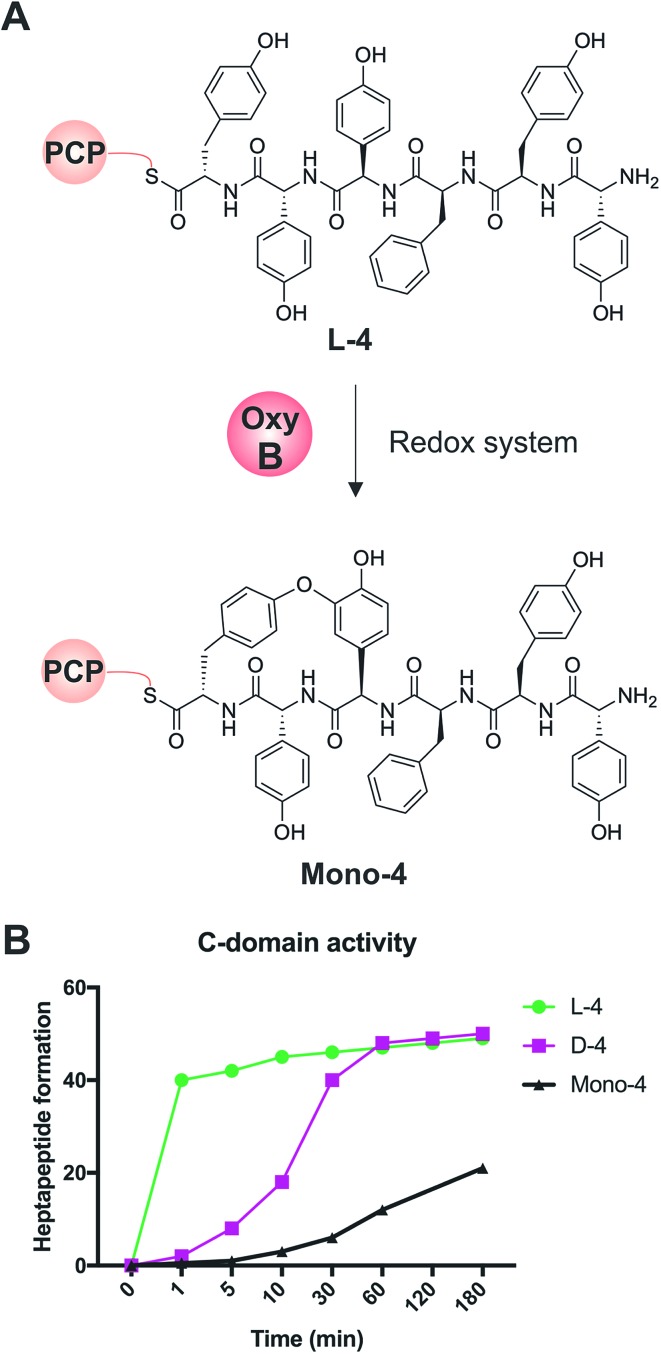
Non-ribosomal peptide synthesis is a highly important biosynthetic pathway for the formation of many secondary metabolites of medical relevance. Due to the challenges associated with the chemical synthesis of many of the products of these assembly lines, understanding the activity and selectivity of non-ribosomal peptide synthetase (NRPS) machineries is an essential step towards the redesign of such machineries to produce new bioactive peptides. Whilst the selectivity of the adenylation domains responsible for amino acid activation during NRPS synthesis has been widely studied, the selectivity of the essential peptide bond forming domains - known as condensation domains - is not well understood. Here, we present the results of a combination of in vitro and in vivo investigations into the final condensation domain from the NRPS machinery that produces the glycopeptide antibiotics (GPAs). Our results show that this condensation domain is tolerant for a range of peptide substrates and even those with unnatural stereochemistry of the peptide C-terminus, which is in contrast to the widely ascribed role of these domains as a stereochemical gatekeeper during NRPS synthesis. Furthermore, we show that this condensation domain has a significant preference for linear peptide substrates over crosslinked peptides, which indicates that the GPA crosslinking cascade targets the heptapeptide bound to the final module of the NRPS machinery and reinforces the role of the unique GPA X-domain in this process. Finally, we demonstrate that the peptide bond forming activity of this condensation domain is coupled to the rate of amino acid activation performed by the subsequent adenylation domain. This is a significant result with implications for NRPS redesign, as it indicates that the rate of amino acid activation of modified adenylation domains must be maintained to prevent unwanted peptide hydrolysis from the NRPS due to a loss of the productive coupling of amino acid selection and peptide bond formation. Taken together, our results indicate that assessing condensation domain activity is a vital step in not only understanding the biosynthetic logic and timing of NRPS-mediated peptide assembly, but also the rules which redesign efforts must obey in order to successfully produce functional, modified NRPS assembly lines.
Figures
References
-
- Weissman K. J. Nat. Prod. Rep. 2016;33:203–230. - PubMed
-
- Süssmuth R. D., Mainz A. Angew. Chem., Int. Ed. 2017;56:3770–3821. - PubMed
-
- Winn M., Fyans J. K., Zhuo Y., Micklefield J. Nat. Prod. Rep. 2016;33:317–347. - PubMed
-
- Payne J. A. E., Schoppet M., Hansen M. H., Cryle M. J. Mol. BioSyst. 2017;13:9–22. - PubMed
LinkOut - more resources
Full Text Sources