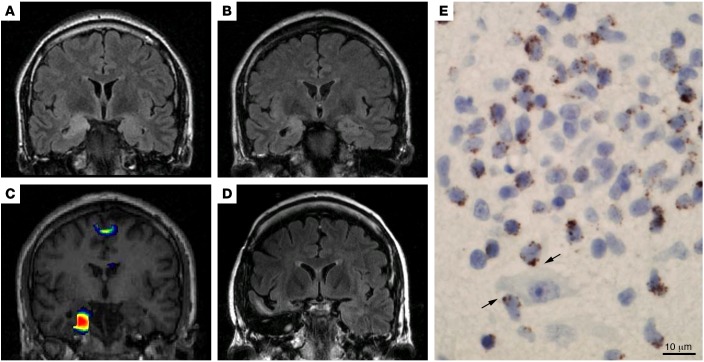
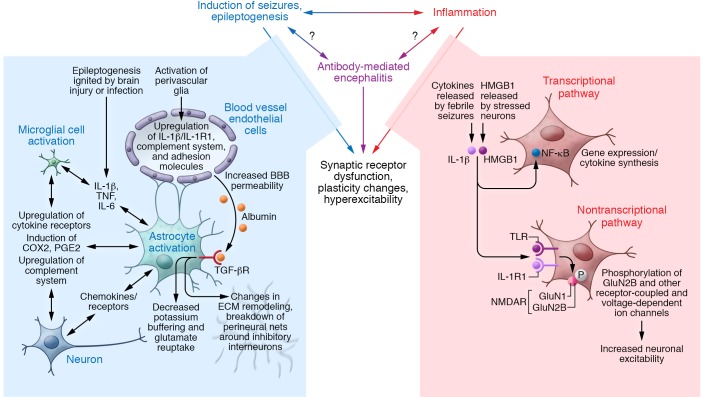
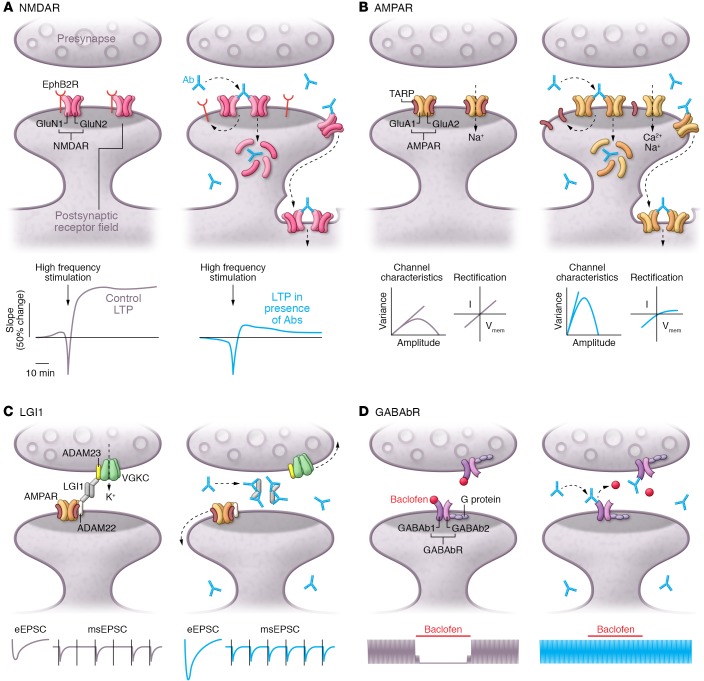
Autoimmune seizures and epilepsy
- PMID: 30714986
- PMCID: PMC6391086
- DOI: 10.1172/JCI125178
Autoimmune seizures and epilepsy
Abstract
The rapid expansion in the number of encephalitis disorders associated with autoantibodies against neuronal proteins has led to an incremental increase in use of the term "autoimmune epilepsy," yet has occurred with limited attention to the physiopathology of each disease and genuine propensity to develop epilepsy. Indeed, most autoimmune encephalitides present with seizures, but the probability of evolving to epilepsy is relatively small. The risk of epilepsy is higher for disorders in which the antigens are intracellular (often T cell-mediated) compared with disorders in which the antigens are on the cell surface (antibody-mediated). Most autoantibodies against neuronal surface antigens show robust effects on the target proteins, resulting in hyperexcitability and impairment of synaptic function and plasticity. Here, we trace the evolution of the concept of autoimmune epilepsy and examine common inflammatory pathways that might lead to epilepsy. Then, we focus on several antibody-mediated encephalitis disorders that associate with seizures and review the synaptic alterations caused by patients' antibodies, with emphasis on those that have been modeled in animals (e.g., antibodies against NMDA, AMPA receptors, LGI1 protein) or in cultured neurons (e.g., antibodies against the GABAb receptor).
Conflict of interest statement
Figures