

Mapping DNA sequence to transcription factor binding energy in vivo
- PMID: 30716072
- PMCID: PMC6375646
- DOI: 10.1371/journal.pcbi.1006226
Mapping DNA sequence to transcription factor binding energy in vivo
Abstract
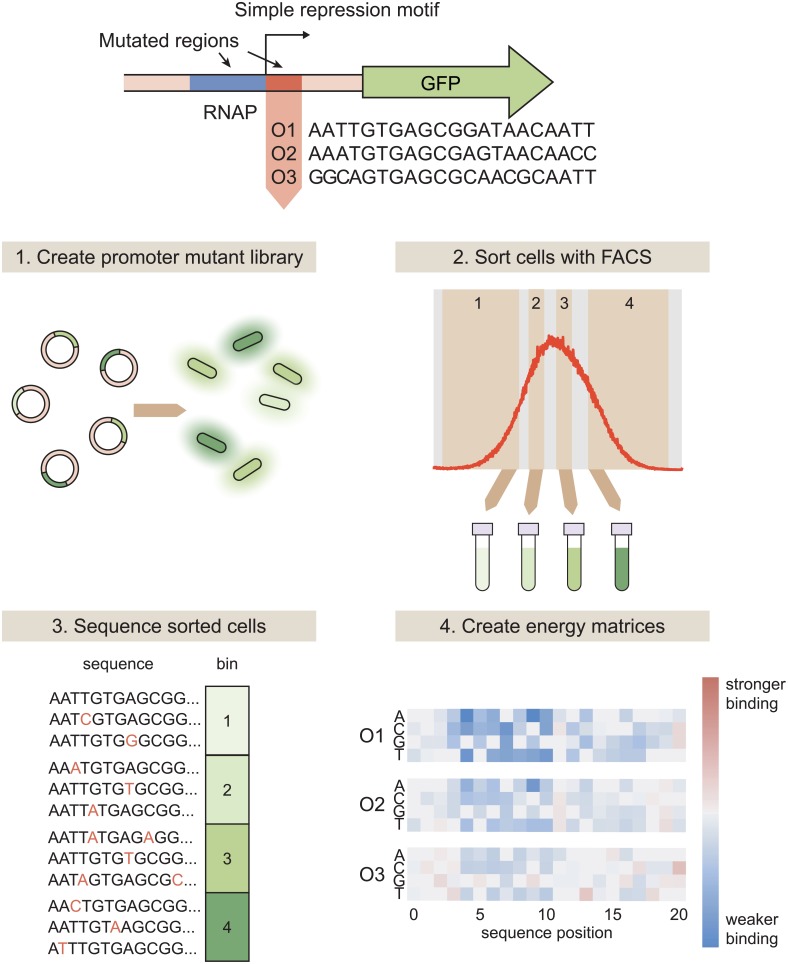
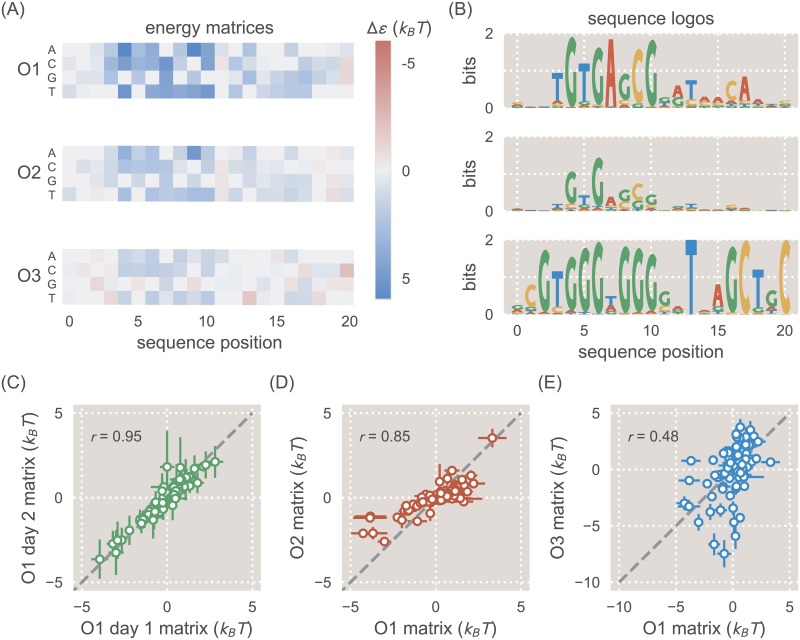
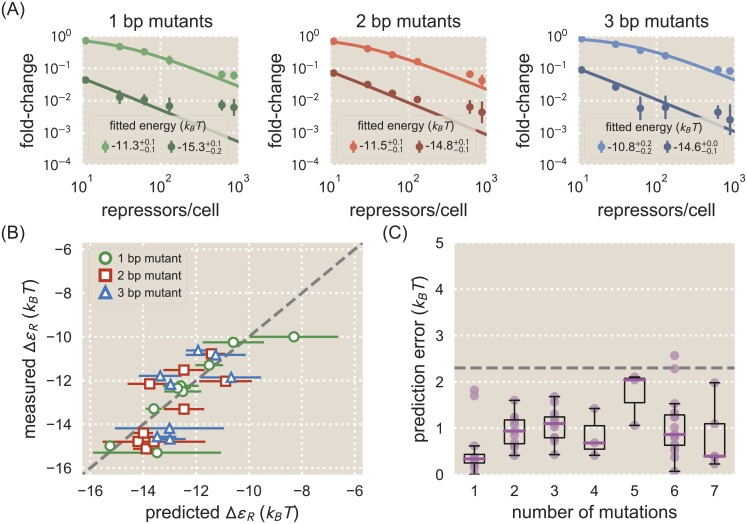
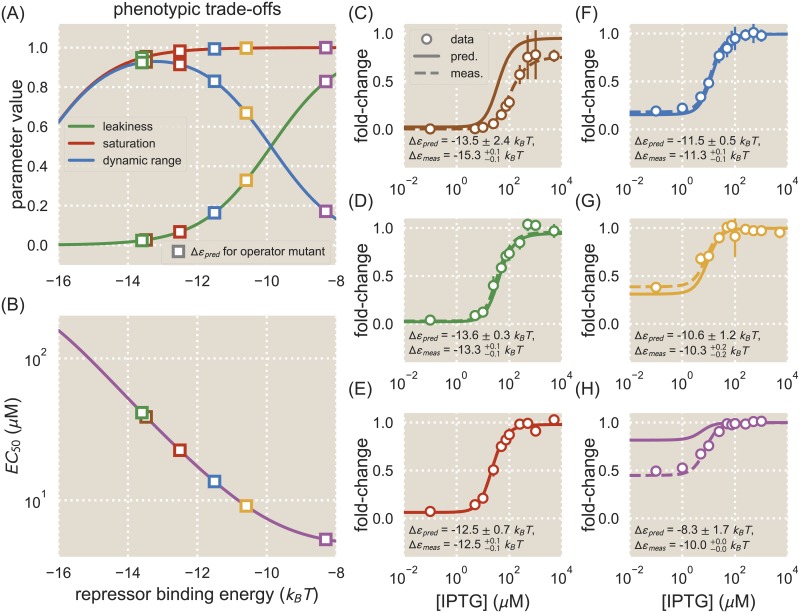
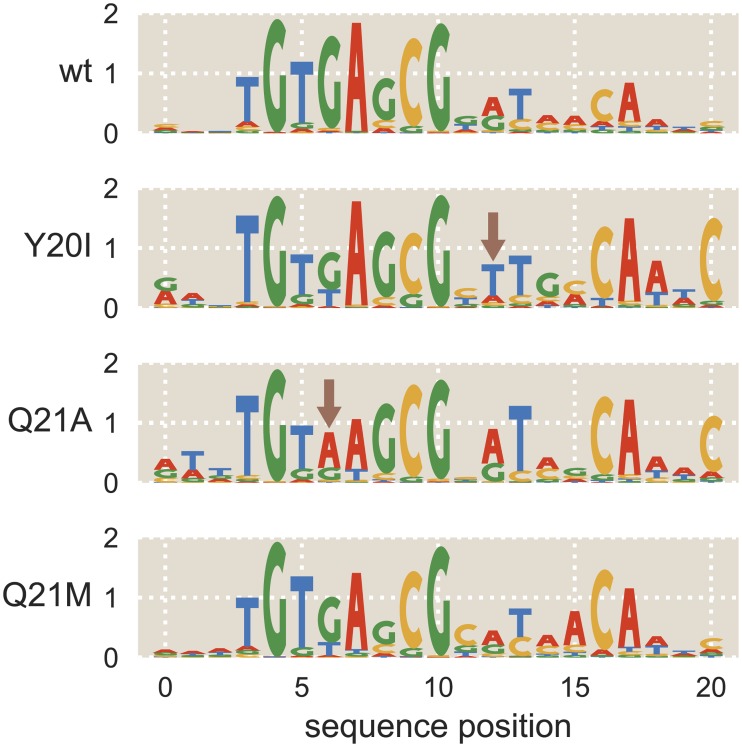
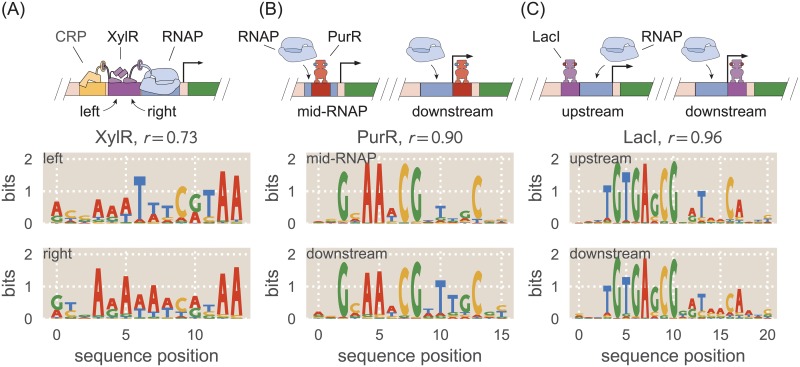
Despite the central importance of transcriptional regulation in biology, it has proven difficult to determine the regulatory mechanisms of individual genes, let alone entire gene networks. It is particularly difficult to decipher the biophysical mechanisms of transcriptional regulation in living cells and determine the energetic properties of binding sites for transcription factors and RNA polymerase. In this work, we present a strategy for dissecting transcriptional regulatory sequences using in vivo methods (massively parallel reporter assays) to formulate quantitative models that map a transcription factor binding site's DNA sequence to transcription factor-DNA binding energy. We use these models to predict the binding energies of transcription factor binding sites to within 1 kBT of their measured values. We further explore how such a sequence-energy mapping relates to the mechanisms of trancriptional regulation in various promoter contexts. Specifically, we show that our models can be used to design specific induction responses, analyze the effects of amino acid mutations on DNA sequence preference, and determine how regulatory context affects a transcription factor's sequence specificity.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
