

Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated with Disease Progression
- PMID: 30718356
- PMCID: PMC6659117
- DOI: 10.1158/1078-0432.CCR-18-3309
Single-Cell Profiling of Cutaneous T-Cell Lymphoma Reveals Underlying Heterogeneity Associated with Disease Progression
Abstract
Purpose: Cutaneous T-cell lymphomas (CTCL), encompassing a spectrum of T-cell lymphoproliferative disorders involving the skin, have collectively increased in incidence over the last 40 years. Sézary syndrome is an aggressive form of CTCL characterized by significant presence of malignant cells in both the blood and skin. The guarded prognosis for Sézary syndrome reflects a lack of reliably effective therapy, due, in part, to an incomplete understanding of disease pathogenesis.
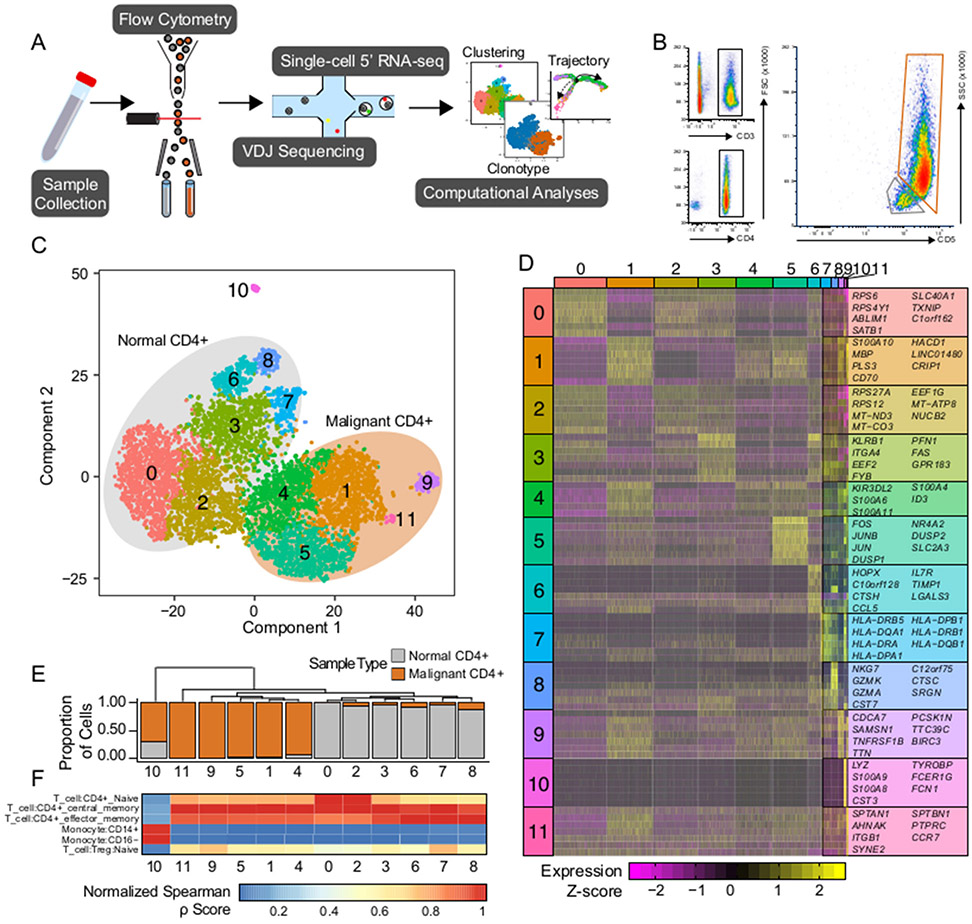
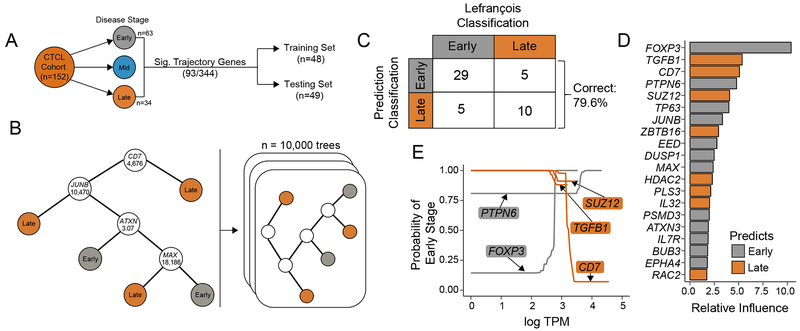
Experimental design: Using single-cell sequencing of RNA and the machine-learning reverse graph embedding approach in the Monocle package, we defined a model featuring distinct transcriptomic states within Sézary syndrome. Gene expression used to differentiate the unique transcriptional states were further used to develop a boosted tree classification for early versus late CTCL disease.
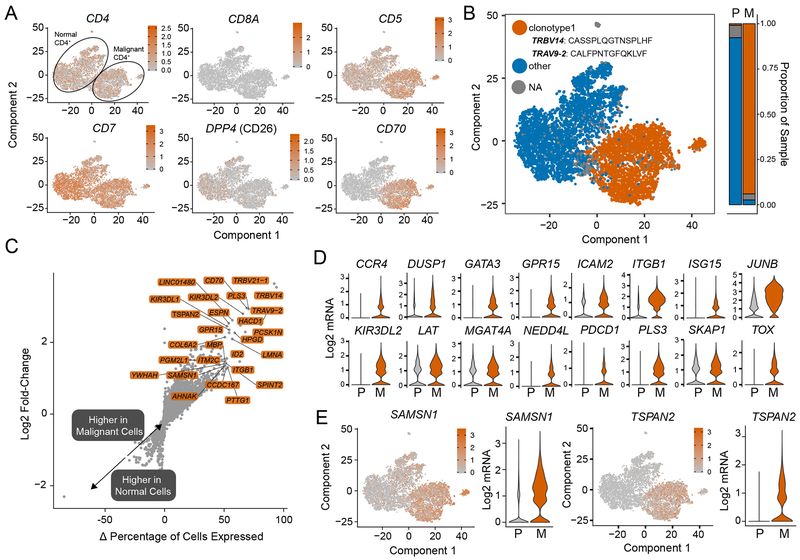
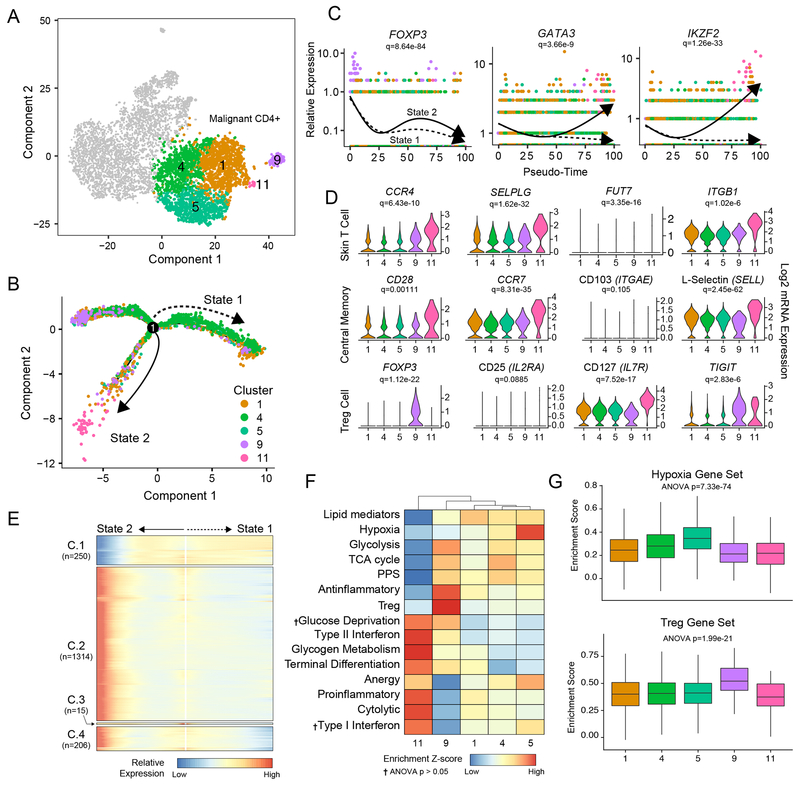
Results: Our analysis showed the involvement of FOXP3 + malignant T cells during clonal evolution, transitioning from FOXP3 + T cells to GATA3 + or IKZF2 + (HELIOS) tumor cells. Transcriptomic diversities in a clonal tumor can be used to predict disease stage, and we were able to characterize a gene signature that predicts disease stage with close to 80% accuracy. FOXP3 was found to be the most important factor to predict early disease in CTCL, along with another 19 genes used to predict CTCL stage.
Conclusions: This work offers insight into the heterogeneity of Sézary syndrome, providing better understanding of the transcriptomic diversities within a clonal tumor. This transcriptional heterogeneity can predict tumor stage and thereby offer guidance for therapy.
©2019 American Association for Cancer Research.
Conflict of interest statement
Declaration of Interests
The authors declare no competing interests
Figures
References
-
- Willemze R, Jaffe ES, Burg G, Cerroni L, Berti E, Swerdlow SH, et al. WHO-EORTC classification for cutaneous lymphomas. Blood. 2005. page 3768–85. - PubMed
-
- Ormsby A, Bergfeld WF, Tubbs RR, Hsi ED. Evaluation of a new paraffin-reactive CD7 T-cell deletion marker and a polymerase chain reaction-based T-cell receptor gene rearrangement assay: Implications for diagnosis of mycosis fungoides in community clinical practice. J Am Acad Dermatol. 2001;45:405–13. - PubMed
-
- Wilcox RA. Cutaneous T-cell lymphoma: 2017 update on diagnosis, risk-stratification, and management. Am J Hematol. 2017;92:1085–102. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
