

Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology
- PMID: 30718627
- PMCID: PMC6361947
- DOI: 10.1038/s41598-018-37774-2
Modeling spinocerebellar ataxias 2 and 3 with iPSCs reveals a role for glutamate in disease pathology
Abstract
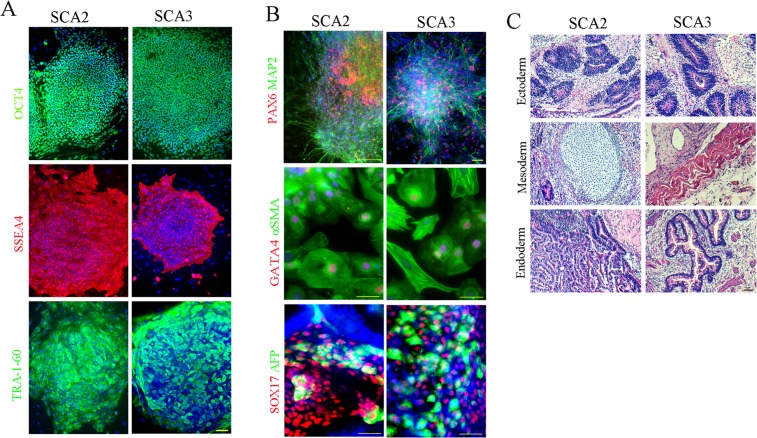
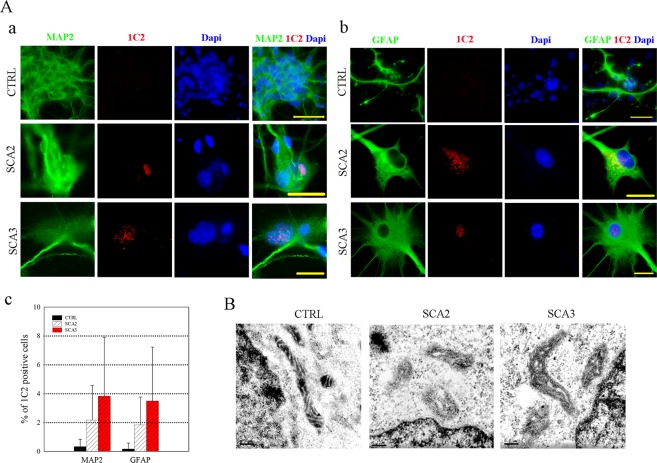
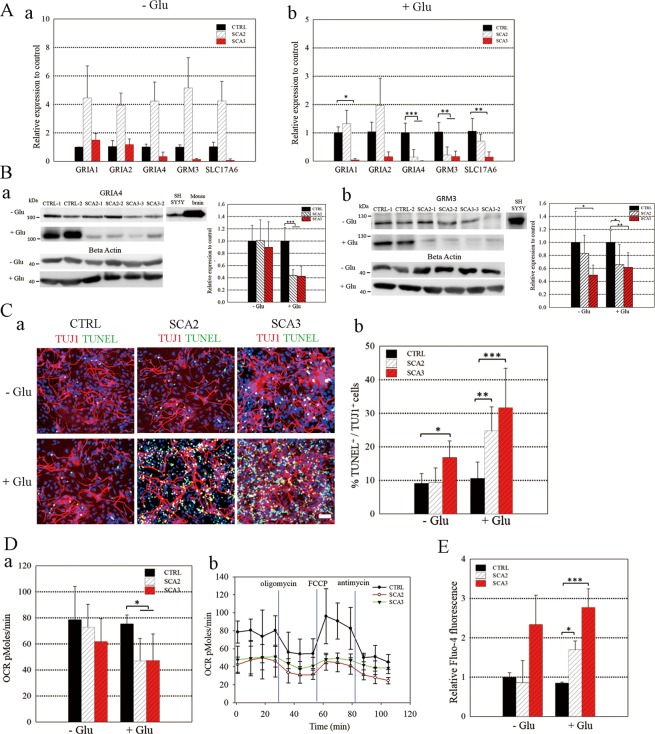
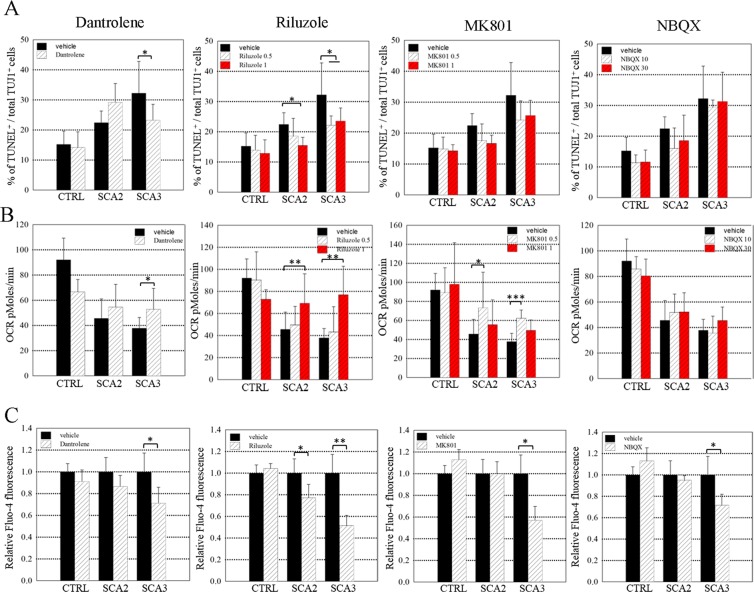
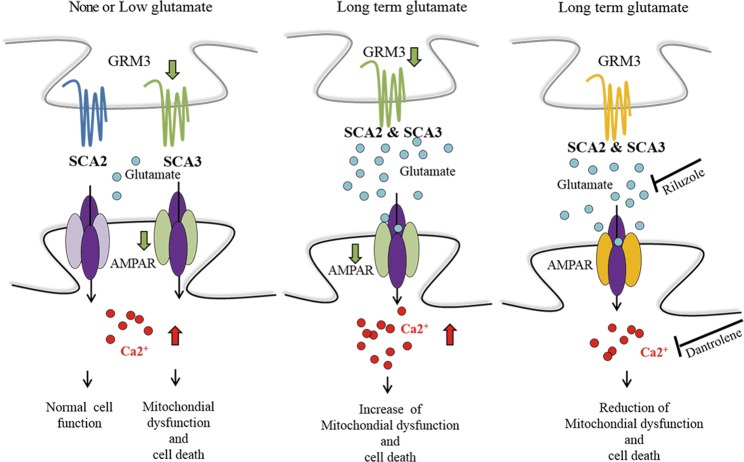
Spinocerebellar ataxias 2 and 3 (SCA2 and SCA3) are dominantly inherited neurodegenerative diseases caused by expansion of polyglutamine-encoding CAG repeats in the affected genes. The etiology of these disorders is known to involve widespread loss of neuronal cells in the cerebellum, however, the mechanisms that contribute to cell death are still elusive. Here we established SCA2 and SCA3 induced pluripotent stem cells (iPSCs) and demonstrated that SCA-associated pathological features can be recapitulated in SCA-iPSC-derived neurons. Importantly, our results also revealed that glutamate stimulation promotes the development of disease-related phenotypes in SCA-iPSC-derived neurons, including altered composition of glutamatergic receptors, destabilized intracellular calcium, and eventual cell death. Furthermore, anti-glutamate drugs and calcium stabilizer treatment protected the SCA-iPSC-derived neurons and reduced cell death. Collectively, our study demonstrates that the SCA-iPSC-derived neurons can recapitulate SCA-associated pathological features, providing a valuable tool to explore SCA pathogenic mechanisms and screen drugs to identify potential SCA therapeutics.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
