

1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses
- PMID: 30718868
- PMCID: PMC6784896
- DOI: 10.1038/s41587-018-0008-8
1,520 reference genomes from cultivated human gut bacteria enable functional microbiome analyses
Abstract
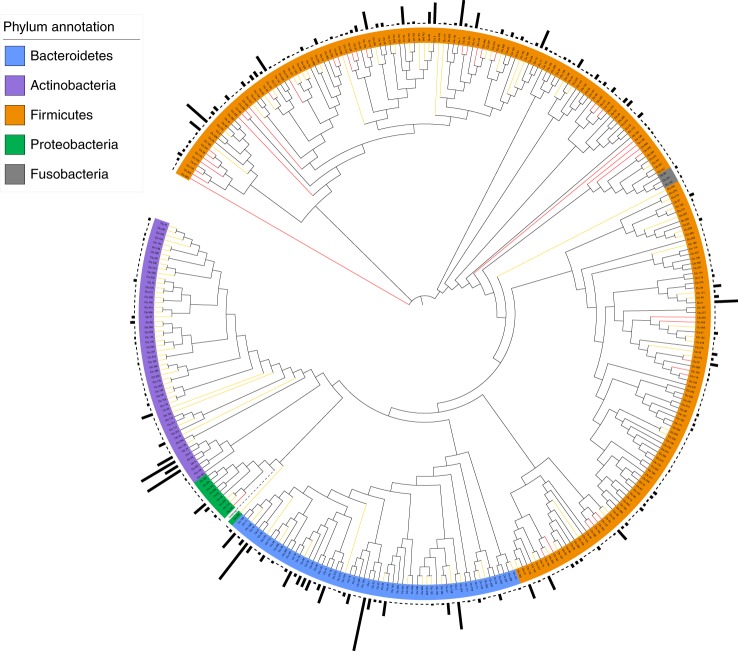
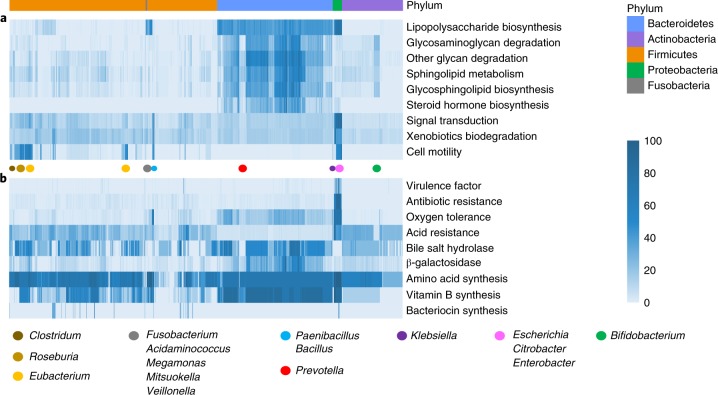
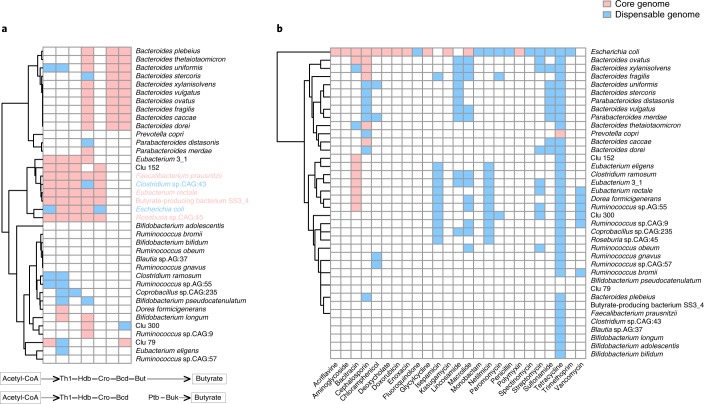
Reference genomes are essential for metagenomic analyses and functional characterization of the human gut microbiota. We present the Culturable Genome Reference (CGR), a collection of 1,520 nonredundant, high-quality draft genomes generated from >6,000 bacteria cultivated from fecal samples of healthy humans. Of the 1,520 genomes, which were chosen to cover all major bacterial phyla and genera in the human gut, 264 are not represented in existing reference genome catalogs. We show that this increase in the number of reference bacterial genomes improves the rate of mapping metagenomic sequencing reads from 50% to >70%, enabling higher-resolution descriptions of the human gut microbiome. We use the CGR genomes to annotate functions of 338 bacterial species, showing the utility of this resource for functional studies. We also carry out a pan-genome analysis of 38 important human gut species, which reveals the diversity and specificity of functional enrichment between their core and dispensable genomes.
Conflict of interest statement
The authors declare no competing interests.
Figures

Comment in
-
Human gut bacterial genome reference.Nat Methods. 2019 Apr;16(4):286. doi: 10.1038/s41592-019-0384-0. Nat Methods. 2019. PMID: 30923389 No abstract available.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
