

RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth
- PMID: 30721156
- PMCID: PMC6436899
- DOI: 10.1172/JCI123049
RABL6A inhibits tumor-suppressive PP2A/AKT signaling to drive pancreatic neuroendocrine tumor growth
Abstract
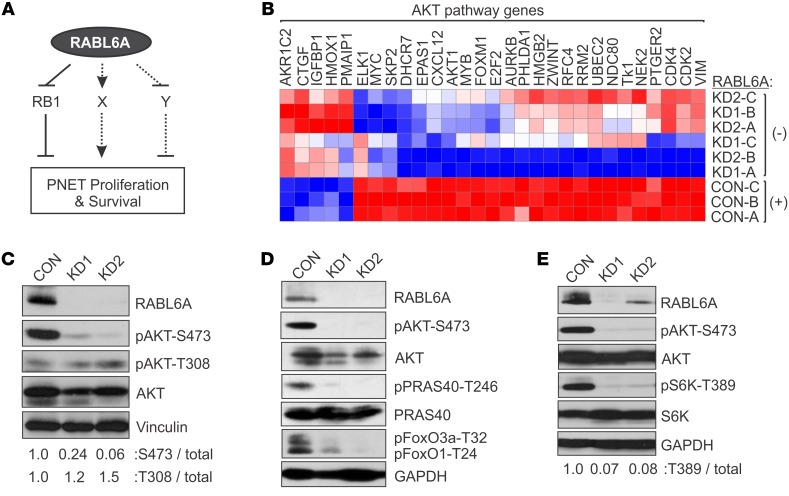
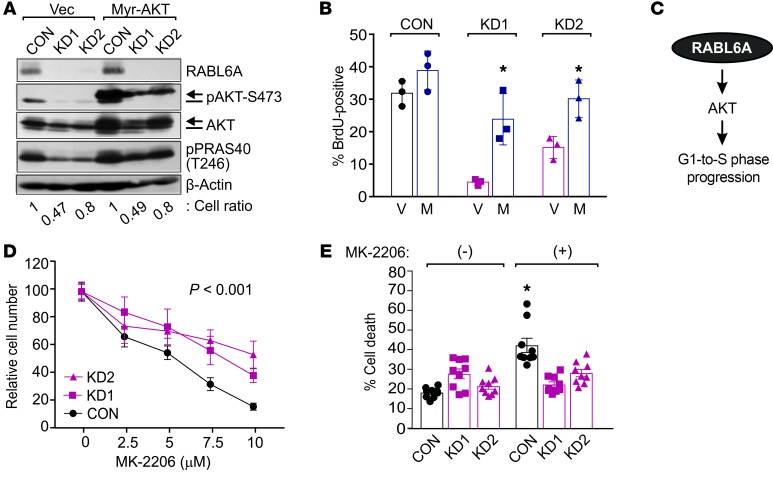
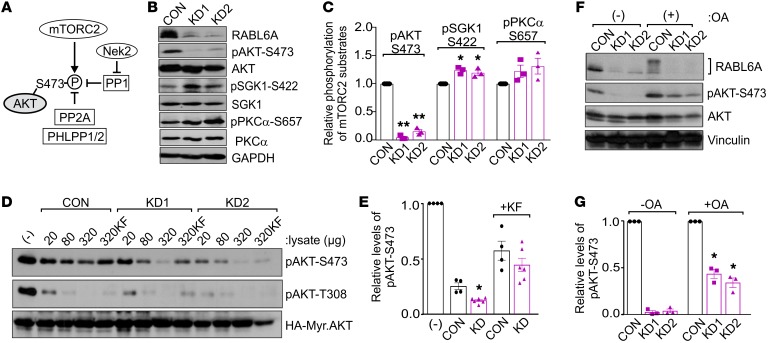
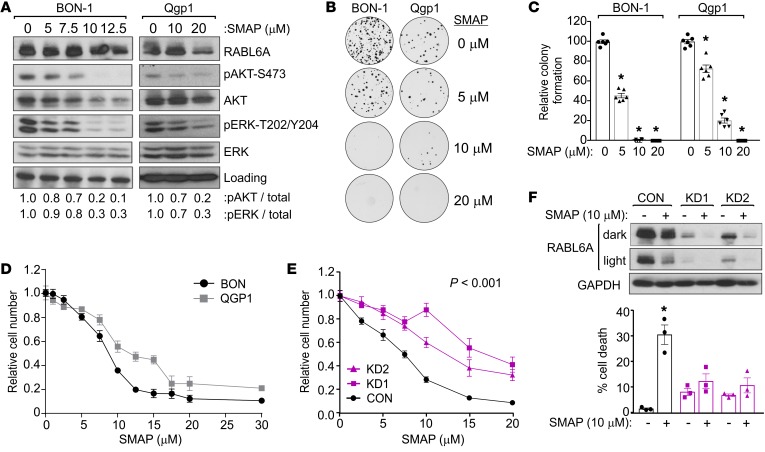
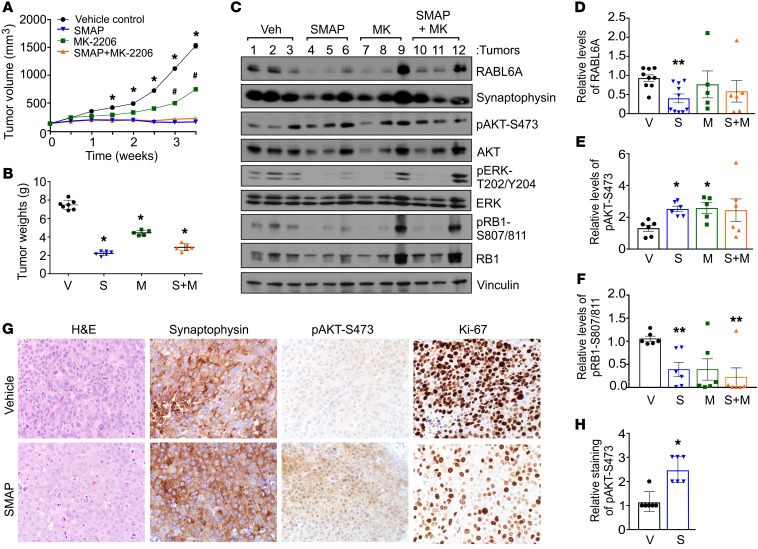
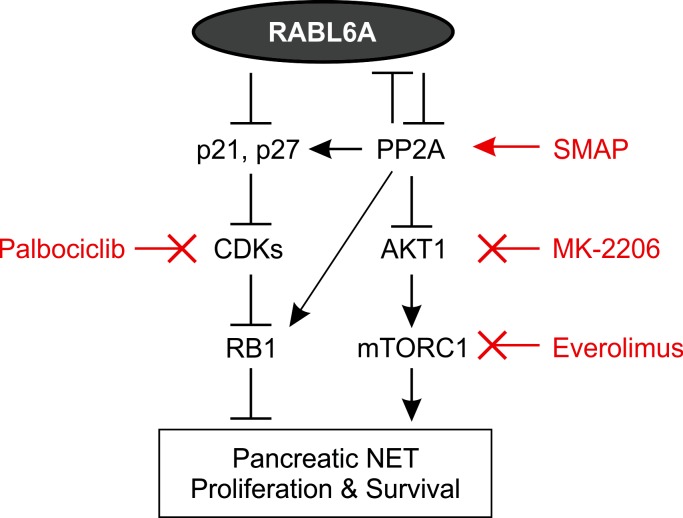
Hyperactivated AKT/mTOR signaling is a hallmark of pancreatic neuroendocrine tumors (PNETs). Drugs targeting this pathway are used clinically, but tumor resistance invariably develops. A better understanding of factors regulating AKT/mTOR signaling and PNET pathogenesis is needed to improve current therapies. We discovered that RABL6A, a new oncogenic driver of PNET proliferation, is required for AKT activity. Silencing RABL6A caused PNET cell-cycle arrest that coincided with selective loss of AKT-S473 (not T308) phosphorylation and AKT/mTOR inactivation. Restoration of AKT phosphorylation rescued the G1 phase block triggered by RABL6A silencing. Mechanistically, loss of AKT-S473 phosphorylation in RABL6A-depleted cells was the result of increased protein phosphatase 2A (PP2A) activity. Inhibition of PP2A restored phosphorylation of AKT-S473 in RABL6A-depleted cells, whereas PP2A reactivation using a specific small-molecule activator of PP2A (SMAP) abolished that phosphorylation. Moreover, SMAP treatment effectively killed PNET cells in a RABL6A-dependent manner and suppressed PNET growth in vivo. The present work identifies RABL6A as a new inhibitor of the PP2A tumor suppressor and an essential activator of AKT in PNET cells. Our findings offer what we believe is a novel strategy of PP2A reactivation for treatment of PNETs as well as other human cancers driven by RABL6A overexpression and PP2A inactivation.
Keywords: Cancer; Cell Biology; Drug therapy; Oncology; Phosphoprotein phosphatases.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
