

Inhibition of Hedgehog signaling reprograms the dysfunctional immune microenvironment in breast cancer
- PMID: 30723576
- PMCID: PMC6350695
- DOI: 10.1080/2162402X.2018.1548241
Inhibition of Hedgehog signaling reprograms the dysfunctional immune microenvironment in breast cancer
Abstract
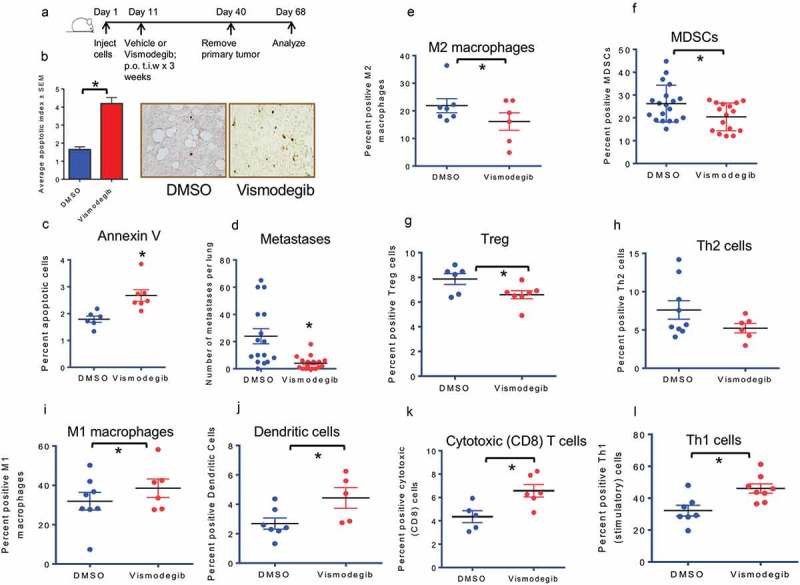
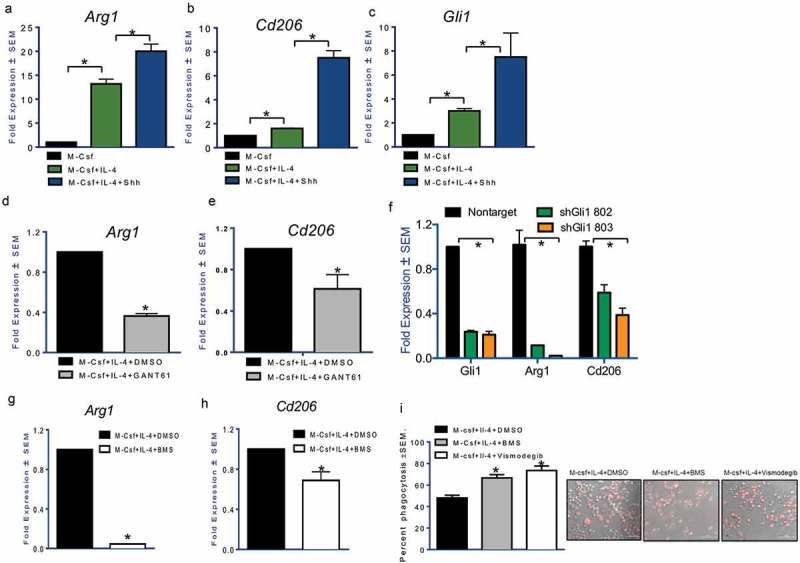
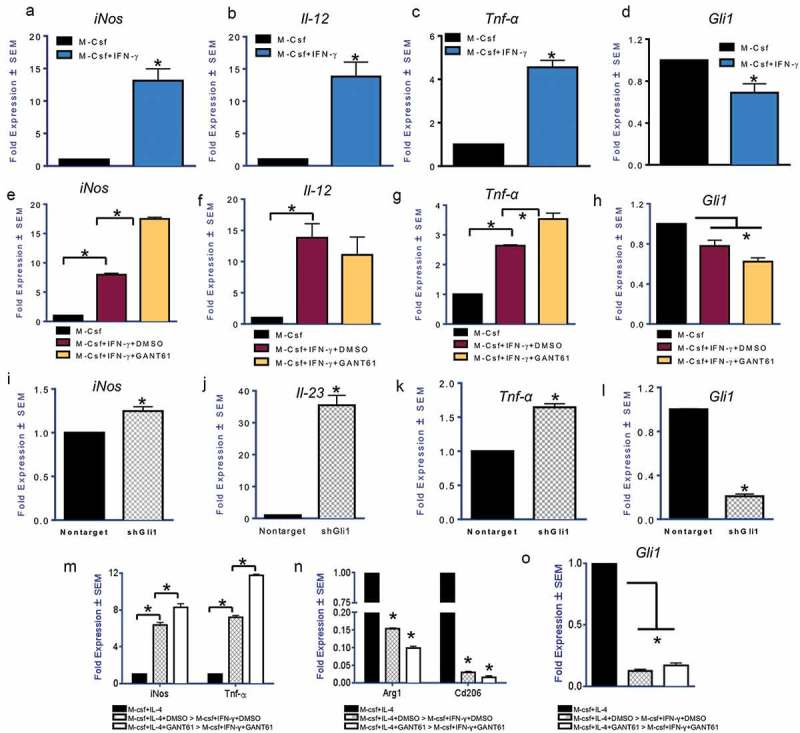
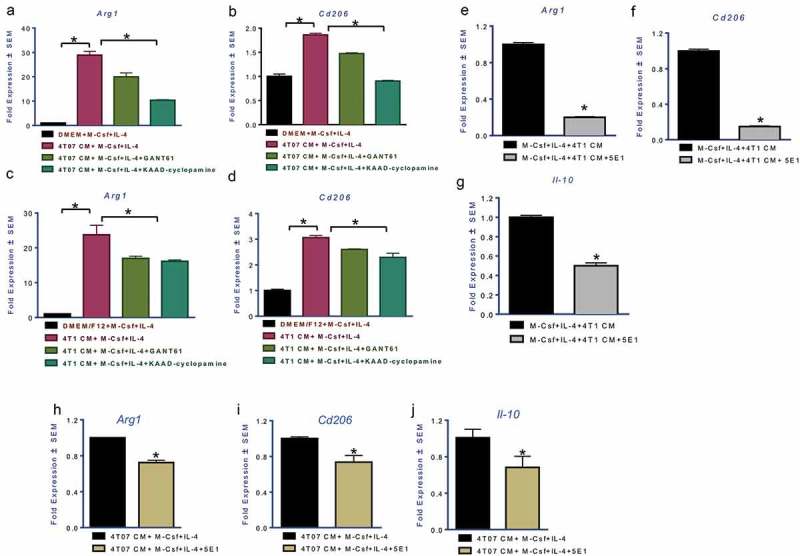
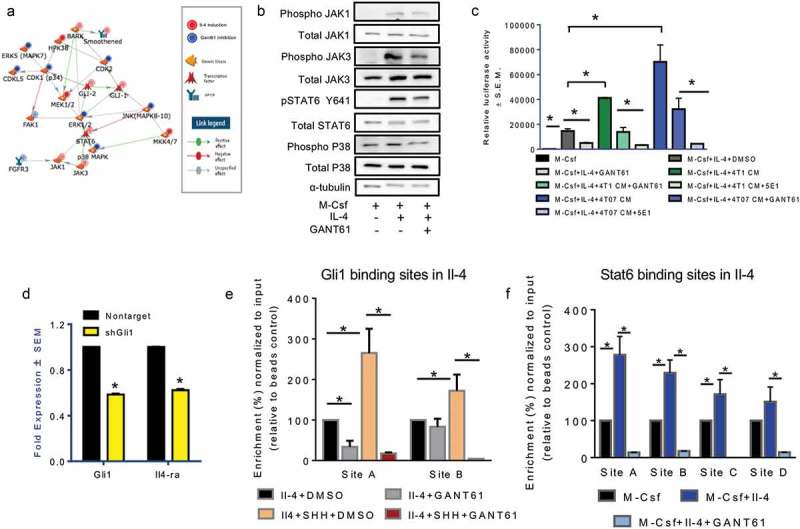
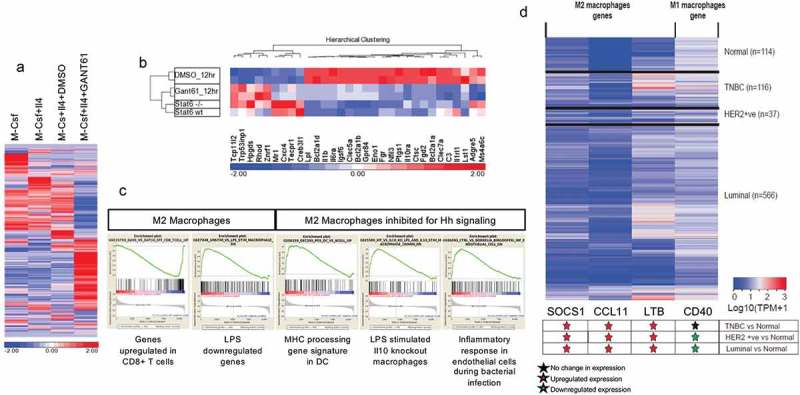
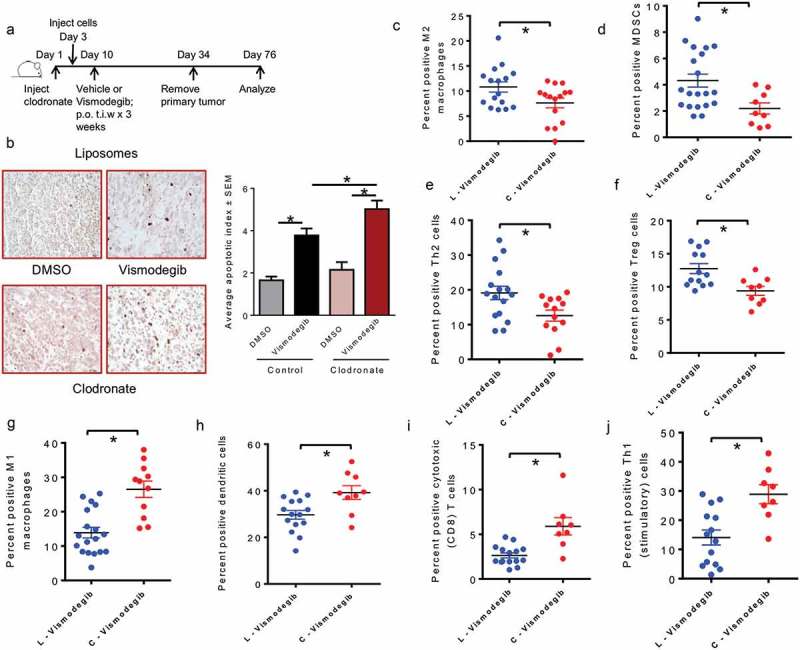
Host responses to tumor cells include tumor suppressing or promoting mechanisms. We sought to detail the effect of Hedgehog (Hh) pathway inhibition on the composition of the mammary tumor immune portfolio. We hypothesized that Hh signaling mediates a crosstalk between breast cancer cells and macrophages that dictates alternative polarization of macrophages and consequently supports a tumor-promoting microenvironment. We used an immunocompetent, syngeneic mouse mammary cancer model to inhibit Hh signaling with the pharmacological inhibitor, Vismodegib. Using molecular and functional assays, we identified that Hedgehog (Hh) signaling mediates a molecular crosstalk between mammary cancer cells and macrophages that culminates in alternative polarization of macrophages. We carried out an unbiased kinomics and genomics assessment to unravel changes in global kinomic and gene signatures impacted by Hh signaling. Our investigations reveal that in an immunocompetent mammary cancer model, the administration of Vismodegib led to changes in the portfolio of tumor-infiltrating immune cells. This was characterized by a marked reduction in immune-suppressive innate and adaptive cells concomitant with an enrichment of cytotoxic immune cells. Breast cancer cells induce M2 polarization of macrophages via a crosstalk mediated by Hh ligands that alters critical kinomic and genomic signatures. Macrophage depletion improved the benefit of Hedgehog inhibition on eliciting an immunogenic, pro-inflammatory profile. We define a novel role for Hh signaling in disabling anti-tumor immunity. Inhibition of Hh signaling presents with dual advantages of tumor cell-targeting as well as re-educating a dysfunctional tumor microenvironment.
Keywords: Hedgehog signaling; breast cancer; metastasis; tumor associated macrophages; tumor microenvironment.
Figures
References
Publication types
Grants and funding
LinkOut - more resources
Full Text Sources