

Emerging links between pediatric lysosomal storage diseases and adult parkinsonism
- PMID: 30726573
- PMCID: PMC6520126
- DOI: 10.1002/mds.27631
Emerging links between pediatric lysosomal storage diseases and adult parkinsonism
Abstract
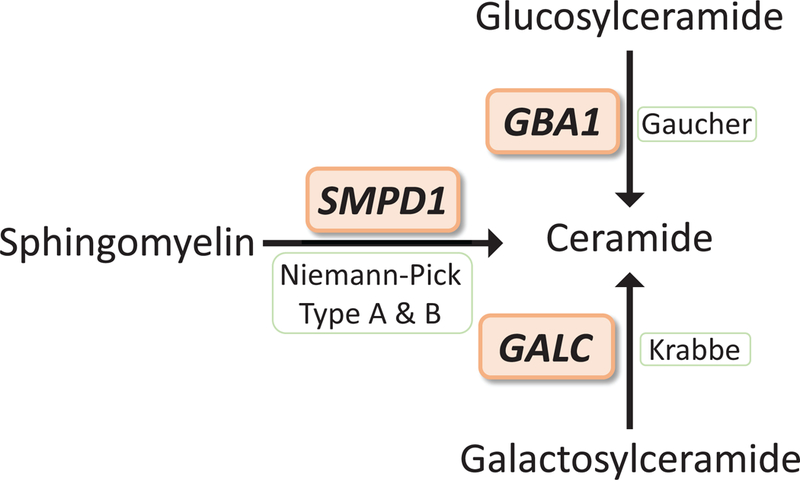
Lysosomal storage disorders comprise a clinically heterogeneous group of autosomal-recessive or X-linked genetic syndromes caused by disruption of lysosomal biogenesis or function resulting in accumulation of nondegraded substrates. Although lysosomal storage disorders are diagnosed predominantly in children, many show variable expressivity with clinical presentations possible later in life. Given the important role of lysosomes in neuronal homeostasis, neurological manifestations, including movement disorders, can accompany many lysosomal storage disorders. Over the last decade, evidence from genetics, clinical epidemiology, cell biology, and biochemistry have converged to implicate links between lysosomal storage disorders and adult-onset movement disorders. The strongest evidence comes from mutations in Glucocerebrosidase, which cause Gaucher's disease and are among the most common and potent risk factors for PD. However, recently, many additional lysosomal storage disorder genes have been similarly implicated, including SMPD1, ATP13A2, GALC, and others. Examination of these links can offer insight into pathogenesis of PD and guide development of new therapeutic strategies. We systematically review the emerging genetic links between lysosomal storage disorders and PD. © 2019 International Parkinson and Movement Disorder Society.
Keywords: Parkinson's disease; genetics; lysosomal storage disease; movement disorders.
© 2019 International Parkinson and Movement Disorder Society.
Conflict of interest statement
Figures

References
-
- Shachar T, Lo Bianco C, Recchia A, Wiessner C, Raas-Rothschild A, Futerman AH. Lysosomal storage disorders and Parkinson’s disease: Gaucher disease and beyond. Mov Disord. 2011;26(9):1593–604. - PubMed
-
- Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat. 2008;29(5):567–83. - PubMed
-
- Vaccaro AM, Motta M, Tatti M, Scarpa S, Masuelli L, Bhat M, et al. Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting. Hum Mol Genet. 2010;19(15):2987–97. - PubMed
-
- Sidransky E Gaucher disease: complexity in a “simple” disorder. Mol Genet Metab. 2004;83(1–2):6–15. - PubMed
-
- Kaplan P, Andersson HC, Kacena KA, Yee JD . The clinical and demographic characteristics of nonneuronopathic Gaucher disease in 887 children at diagnosis. Arch Pediatr Adolesc Med. 2006;160(6):603–8. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
