

Microbiome dysbiosis is associated with disease duration and increased inflammatory gene expression in systemic sclerosis skin
- PMID: 30728065
- PMCID: PMC6366065
- DOI: 10.1186/s13075-019-1816-z
Microbiome dysbiosis is associated with disease duration and increased inflammatory gene expression in systemic sclerosis skin
Abstract
Background: Infectious agents have long been postulated to be disease triggers for systemic sclerosis (SSc), but a definitive link has not been found. Metagenomic analyses of high-throughput data allows for the unbiased identification of potential microbiome pathogens in skin biopsies of SSc patients and allows insight into the relationship with host gene expression.
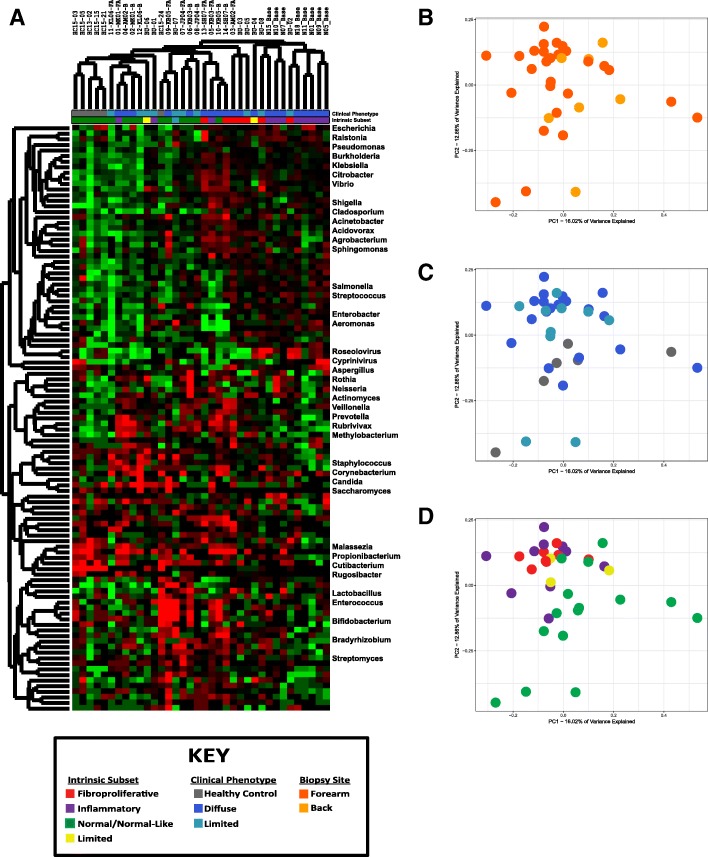
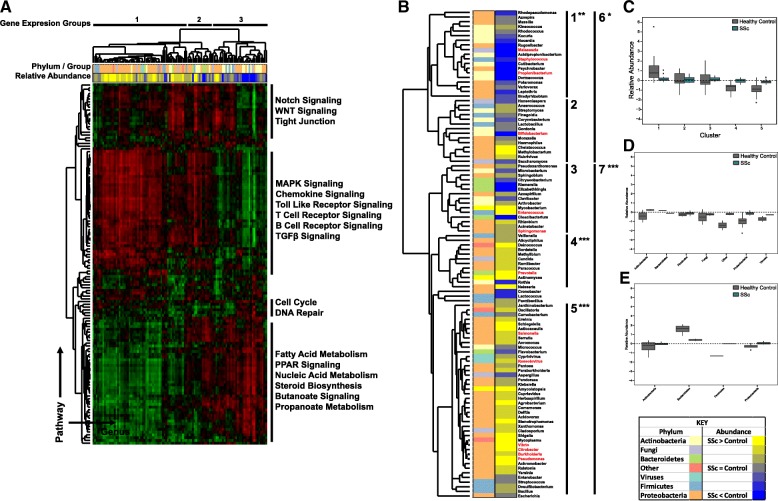
Methods: We examined skin biopsies from a diverse cohort of 23 SSc patients (including lesional forearm and non-lesional back samples) by RNA-seq. Metagenomic filtering and annotation was performed using the Integrated Metagenomic Sequencing Analysis (IMSA). Associations between microbiome composition and gene expression were analyzed using single-sample gene set enrichment analysis (ssGSEA).
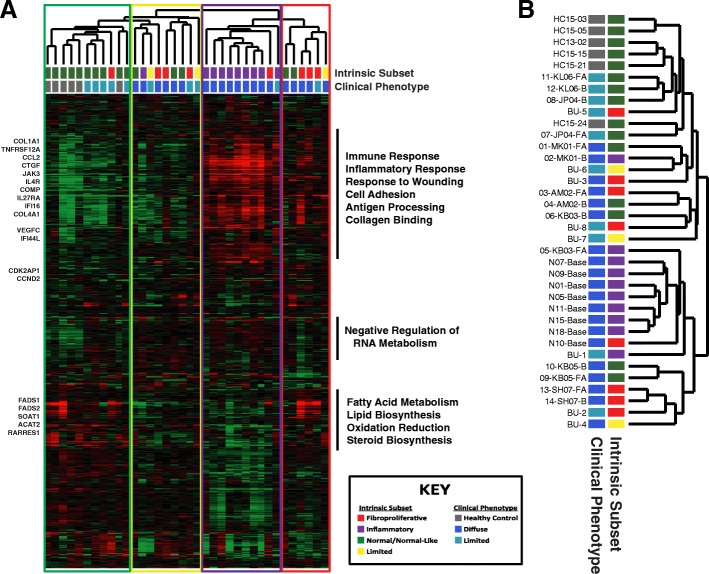
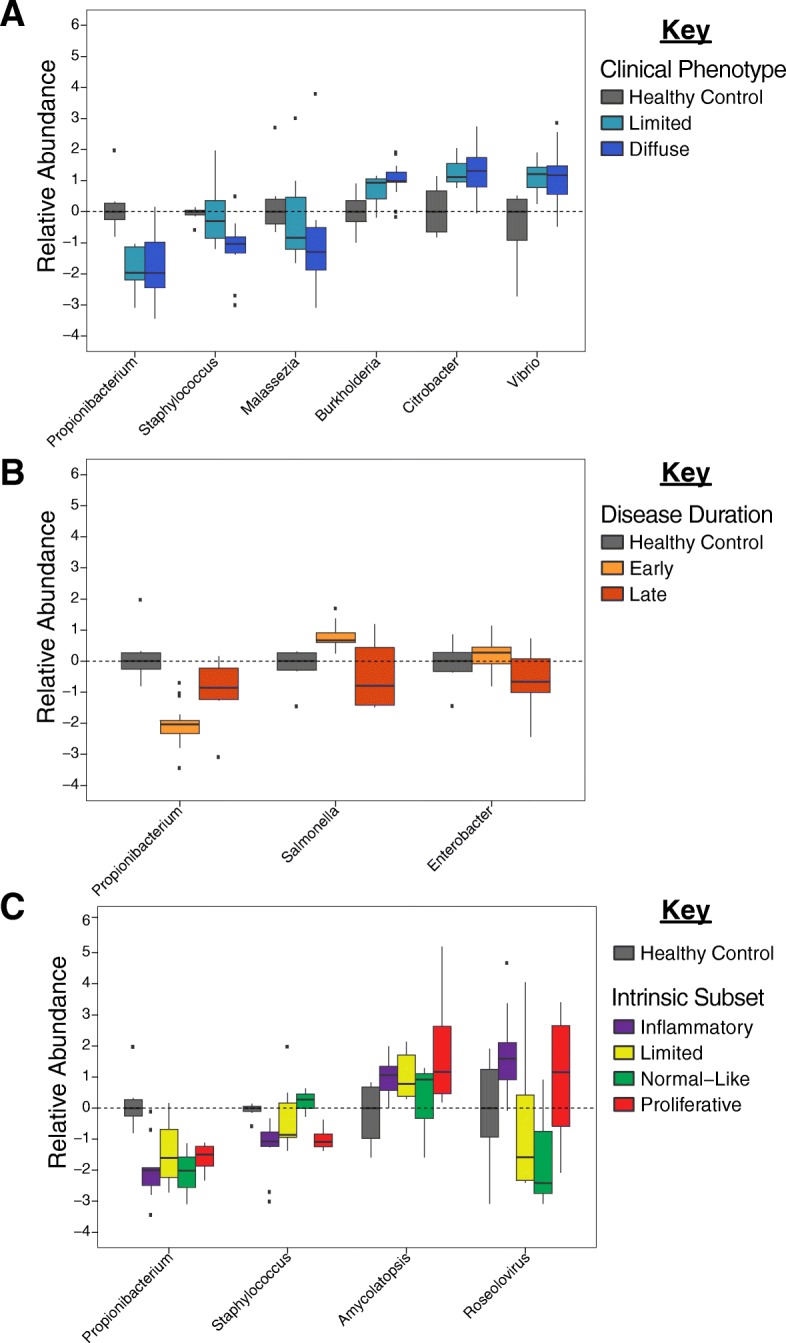
Results: We find the skin of SSc patients exhibits substantial changes in microbial composition relative to controls, characterized by sharp decreases in lipophilic taxa, such as Propionibacterium, combined with increases in a wide range of gram-negative taxa, including Burkholderia, Citrobacter, and Vibrio.
Conclusions: Microbiome dysbiosis is associated with disease duration and increased inflammatory gene expression. These data provide a comprehensive portrait of the SSc skin microbiome and its association with local gene expression, which mirrors the molecular changes in lesional skin.
Keywords: Metagenomics; Microbiome; RNA-sequencing; Scleroderma; Systemic sclerosis.
Conflict of interest statement
Ethics approval and consent to participate
Study participants provided written, informed consent prior to sample collection in accordance with the Declaration of Helsinki Protocol and the Institutional Review Boards of Boston University Medical Center, Boston, MA, Dartmouth-Hitchcock Medical Center, Lebanon, NH, and the Hospital for Special Surgery, New York, NY.
Consent for publication
N/A
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Comment in
-
Cause or effect? Interpreting emerging evidence for dysbiosis in systemic sclerosis.Arthritis Res Ther. 2019 Mar 27;21(1):81. doi: 10.1186/s13075-019-1872-4. Arthritis Res Ther. 2019. PMID: 30917857 Free PMC article.
References
-
- Johnson M, Mahoney J, Taroni J, Sargent J, Marmarelis E, Wu M, Varga J, Hinchcliff M, Whitfield M. Experimentally-derived fibroblast gene signatures identify molecular pathways associated with distinct subsets of systemic sclerosis patients in three independent cohorts. PLoS One. 2015;10(1):e0114017. doi: 10.1371/journal.pone.0114017. - DOI - PMC - PubMed
-
- Mahoney JM, Taroni J, Martyanov V, Wood TA, Greene CS, Pioli PA, Hinchcliff ME, Whitfield ML. Systems level analysis of systemic sclerosis shows a network of immune and profibrotic pathways connected with genetic polymorphisms. PLoS Comput Biol. 2015;11(1):e1004005. doi: 10.1371/journal.pcbi.1004005. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
