

Epidemiological Typing of Serratia marcescens Isolates by Whole-Genome Multilocus Sequence Typing
- PMID: 30728192
- PMCID: PMC6440794
- DOI: 10.1128/JCM.01652-18
Epidemiological Typing of Serratia marcescens Isolates by Whole-Genome Multilocus Sequence Typing
Abstract
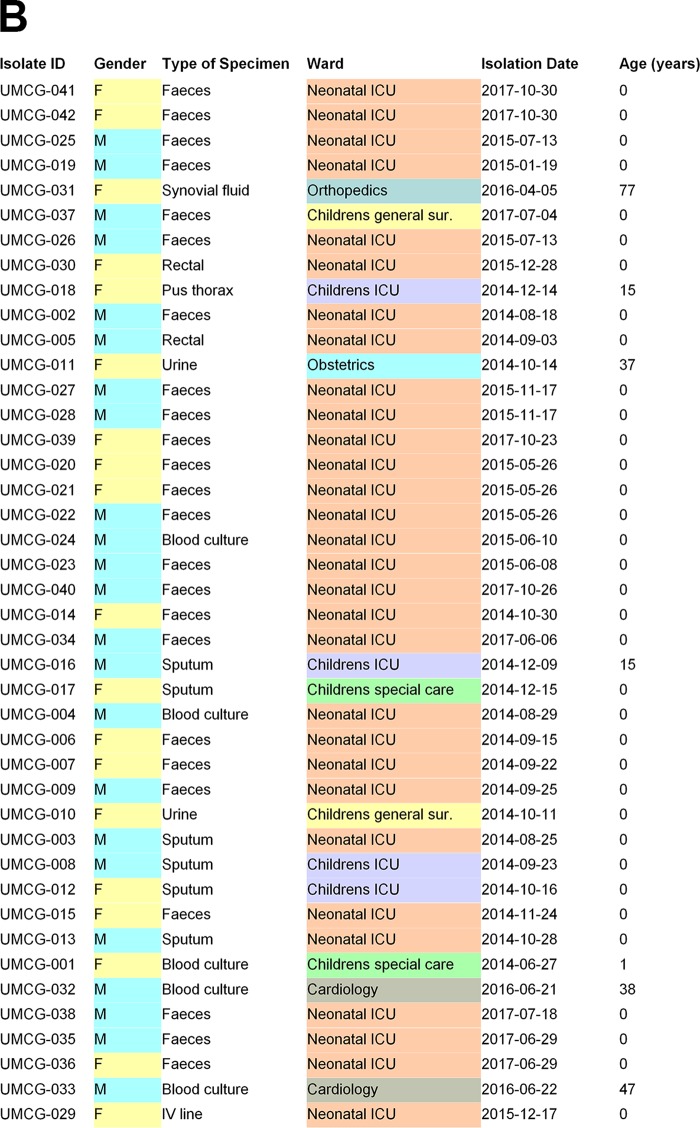
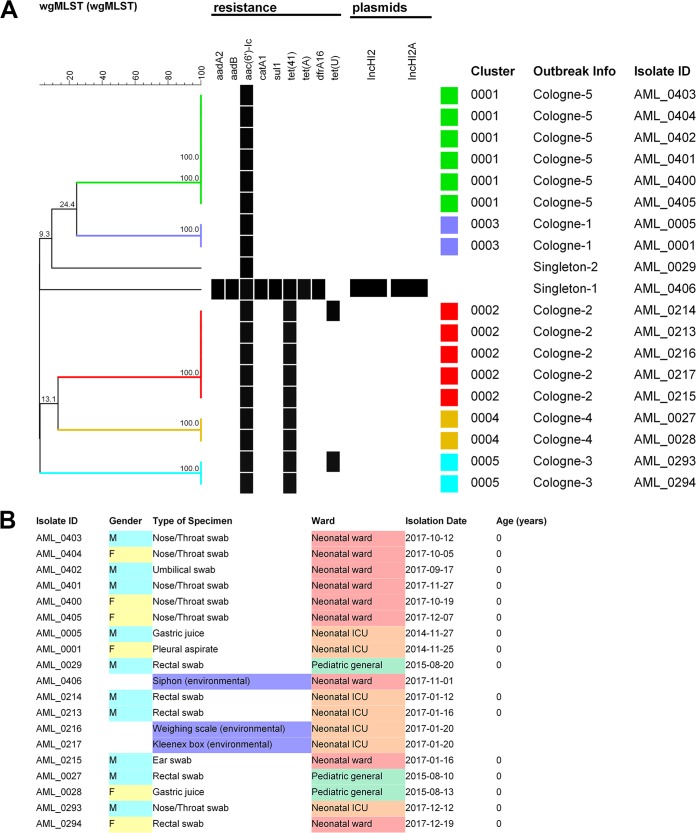
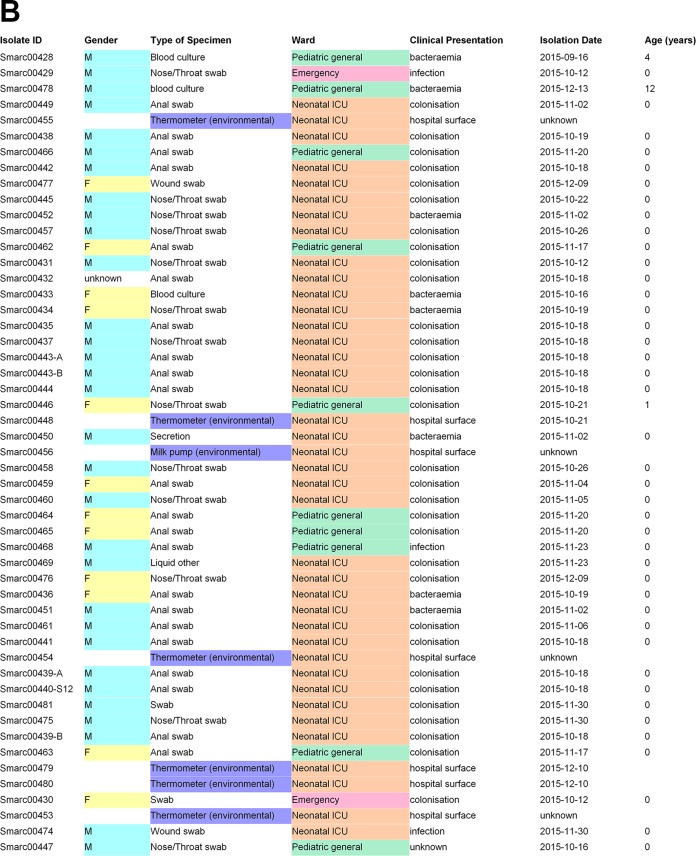
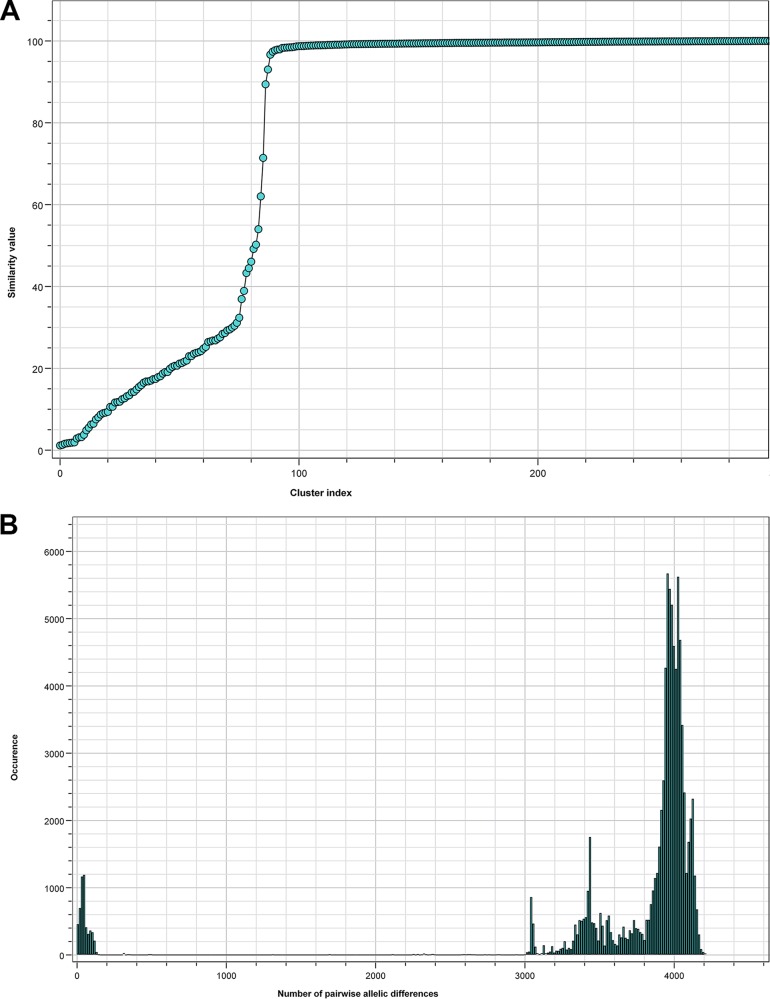
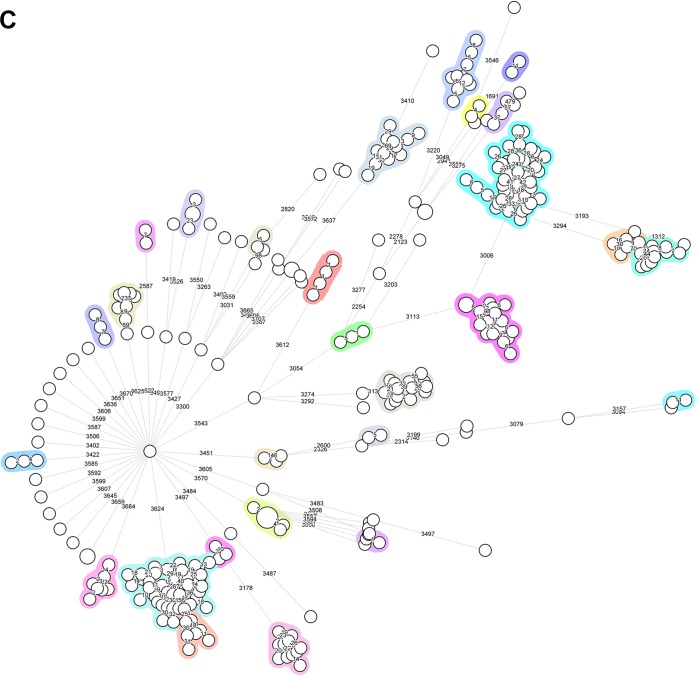
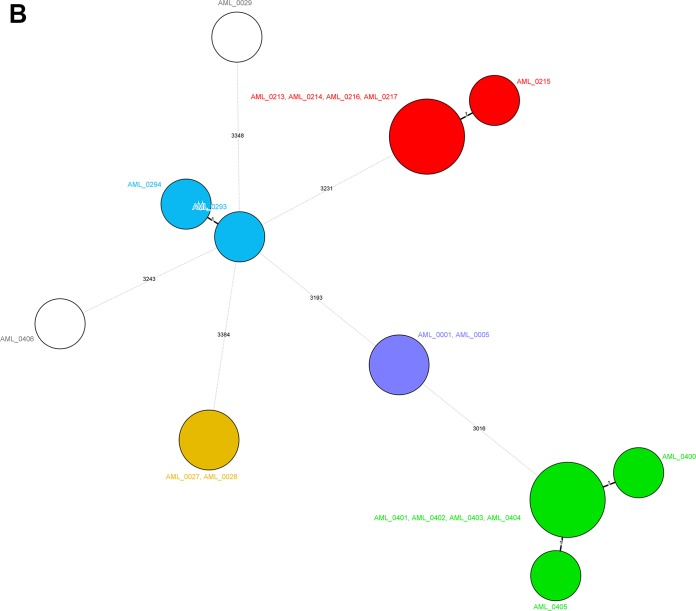
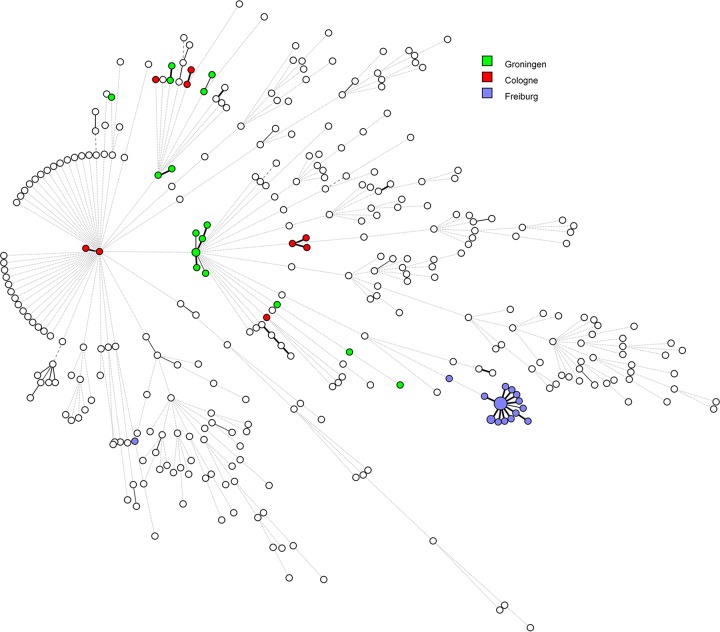
Serratia marcescens is an opportunistic bacterial pathogen. It is notorious for its increasing antimicrobial resistance and its potential to cause outbreaks of colonization and infections, predominantly in neonatal intensive care units (NICUs). There, its spread requires rapid infection control response. To understand its spread, detailed molecular typing is key. We present a whole-genome multilocus sequence typing (wgMLST) method for S. marcescens Using a set of 299 publicly available whole-genome sequences (WGS), we developed an initial wgMLST system consisting of 9,377 gene loci. This included 1,455 loci occurring in all reference genomes and 7,922 accessory loci. This closed system was validated using three geographically diverse collections of S. marcescens consisting of 111 clinical isolates implicated in nosocomial dissemination events in three hospitals. The validation procedure showed a full match between epidemiological data and the wgMLST analyses. We set the cutoff value for epidemiological (non)relatedness at 20 different alleles, though for the majority of outbreak-clustered isolates, this difference was limited to 4 alleles. This shows that the wgMLST system for S. marcescens provides prospects for successful future monitoring for the epidemiological containment of this opportunistic pathogen.
Keywords: Serratia marcescens; WGS; bionumerics; molecular typing; neonatal intensive care; outbreak management; wgMLST.
Copyright © 2019 American Society for Microbiology.
Figures




References
-
- Sabat AJ, Budimir A, Nashev D, Sá-Leão R, van Dijl JM, Laurent F, Grundmann H, Friedrich AW, ESCMID Study Group of Epidemiological Markers (ESGEM). 2013. Overview of molecular typing methods for outbreak detection and epidemiological surveillance. Euro Surveill 18:20380. - PubMed
-
- Goering RV, Köck R, Grundmann H, Werner G, Friedrich AW, ESCMID Study Group for Epidemiological Markers (ESGEM). 2013. From theory to practice: molecular strain typing for the clinical and public health setting. Euro Surveill 18:20383. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
