

Ultra-large library docking for discovering new chemotypes
- PMID: 30728502
- PMCID: PMC6383769
- DOI: 10.1038/s41586-019-0917-9
Ultra-large library docking for discovering new chemotypes
Abstract
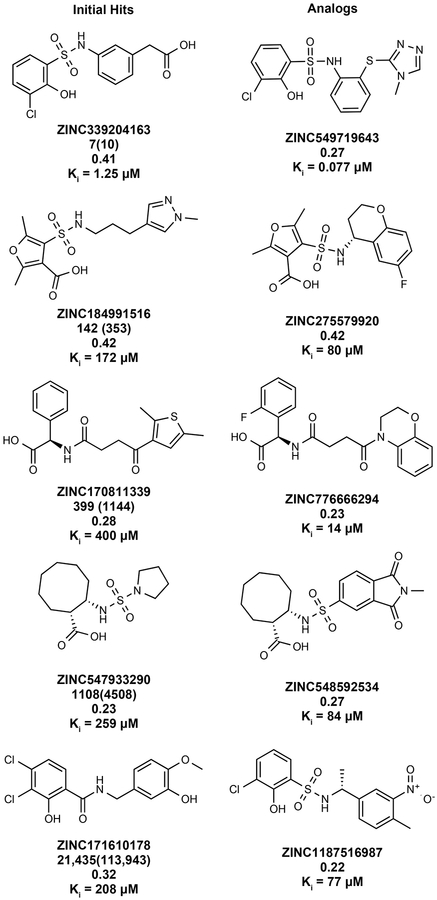
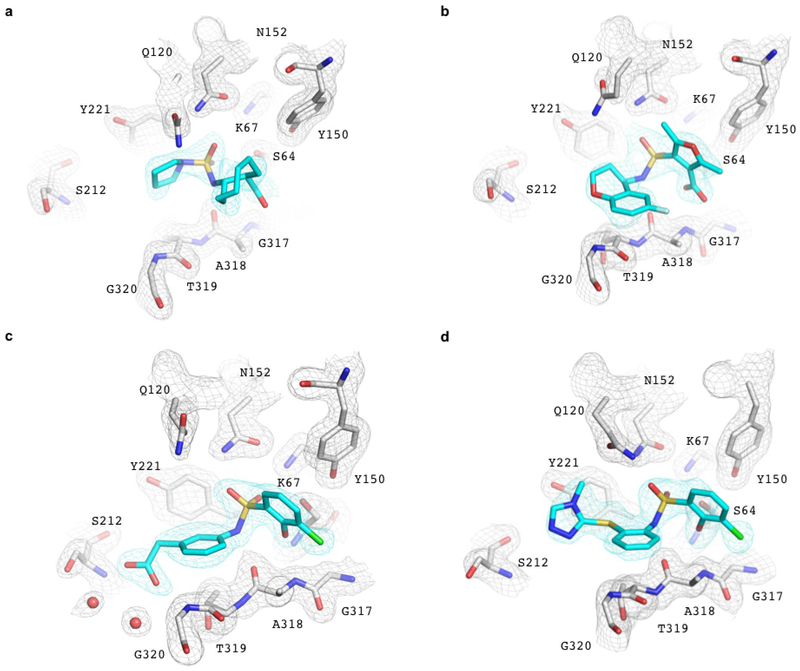
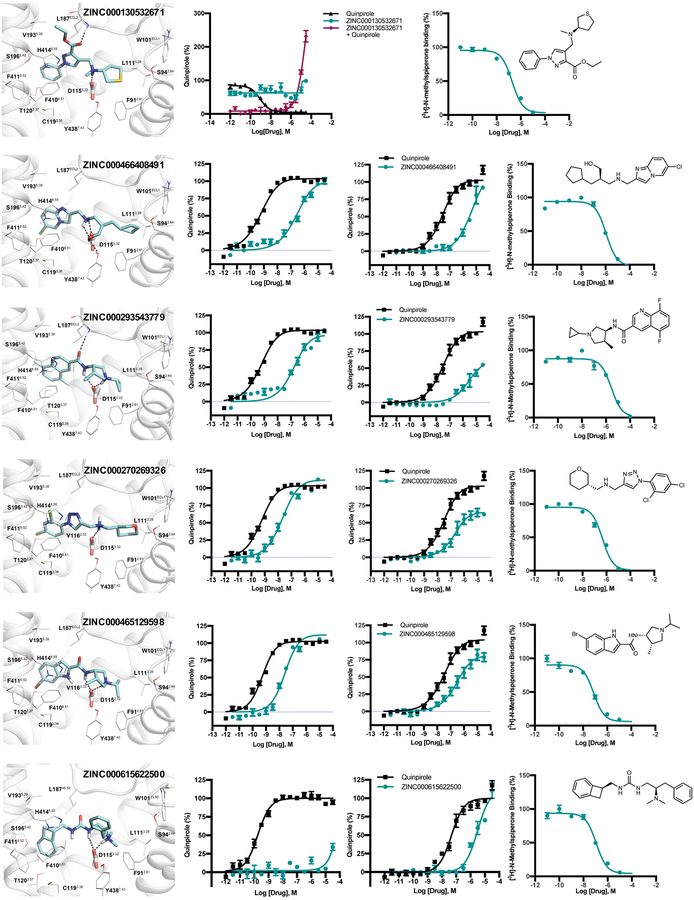
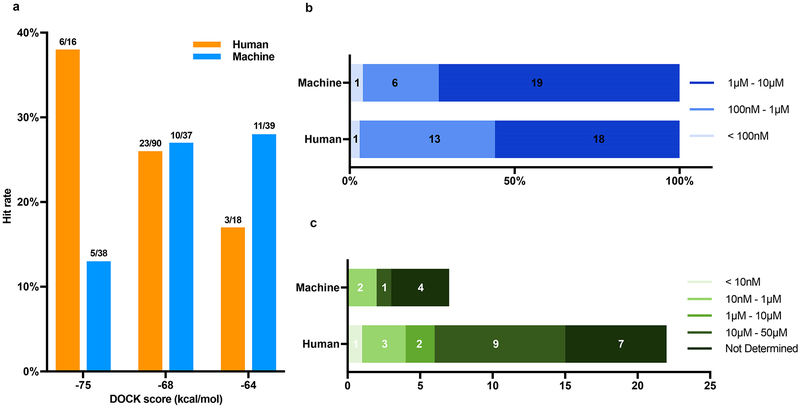
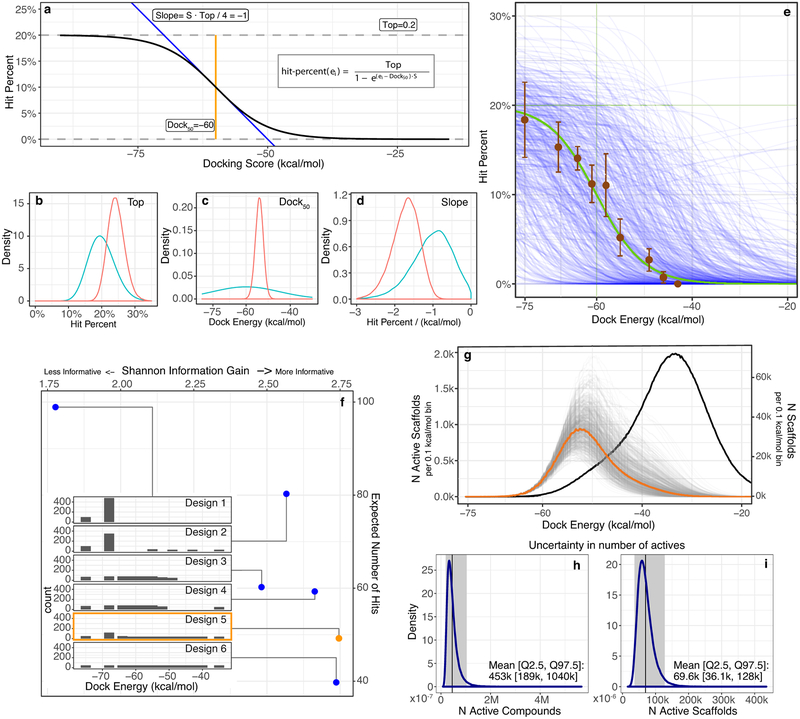
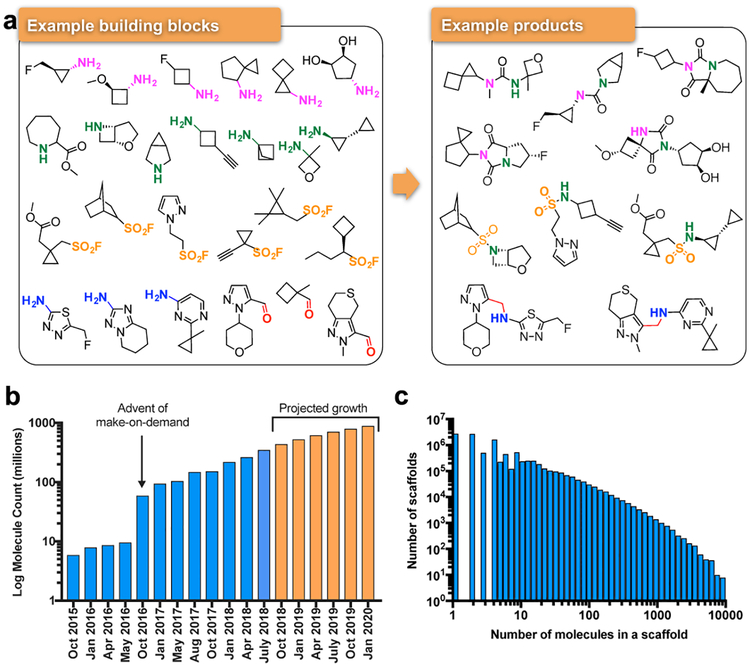
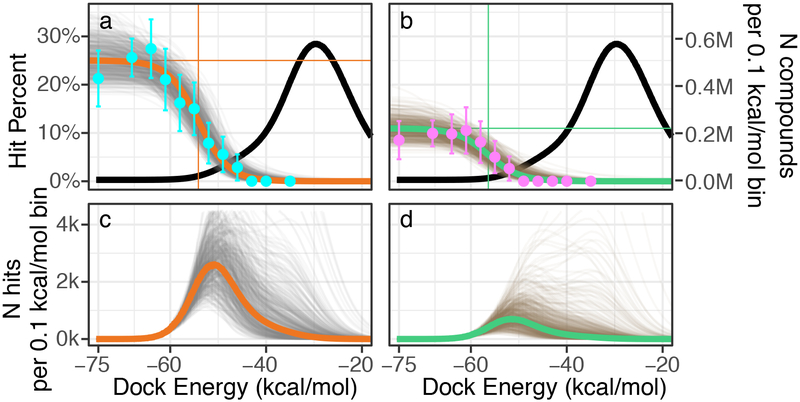
Despite intense interest in expanding chemical space, libraries containing hundreds-of-millions to billions of diverse molecules have remained inaccessible. Here we investigate structure-based docking of 170 million make-on-demand compounds from 130 well-characterized reactions. The resulting library is diverse, representing over 10.7 million scaffolds that are otherwise unavailable. For each compound in the library, docking against AmpC β-lactamase (AmpC) and the D4 dopamine receptor were simulated. From the top-ranking molecules, 44 and 549 compounds were synthesized and tested for interactions with AmpC and the D4 dopamine receptor, respectively. We found a phenolate inhibitor of AmpC, which revealed a group of inhibitors without known precedent. This molecule was optimized to 77 nM, which places it among the most potent non-covalent AmpC inhibitors known. Crystal structures of this and other AmpC inhibitors confirmed the docking predictions. Against the D4 dopamine receptor, hit rates fell almost monotonically with docking score, and a hit-rate versus score curve predicted that the library contained 453,000 ligands for the D4 dopamine receptor. Of 81 new chemotypes discovered, 30 showed submicromolar activity, including a 180-pM subtype-selective agonist of the D4 dopamine receptor.
Conflict of interest statement
Figures





Comment in
-
Bigger is better in virtual drug screens.Nature. 2019 Feb;566(7743):193-194. doi: 10.1038/d41586-019-00145-6. Nature. 2019. PMID: 30737502 No abstract available.
-
Ultra-large virtual molecular libraries throw open chemical space.Nature. 2019 Feb;566(7742):7. doi: 10.1038/d41586-019-00482-6. Nature. 2019. PMID: 30814689 No abstract available.
References
-
- Bohacek RS, McMartin C & Guida WC The art and practice of structure-based drug design: A molecular modeling perspective. Medicinal Research Reviews 16, 3–50, (1996). - PubMed
-
- Ertl P Cheminformatics Analysis of Organic Substituents: Identification of the Most Common Substituents, Calculation of Substituent Properties, and Automatic Identification of Drug-like Bioisosteric Groups. Journal of Chemical Information and Computer Sciences 43, 374–380, (2003). - PubMed
-
- Fink T, Bruggesser H & Reymond JL Virtual Exploration of the Small-Molecule Chemical Universe below 160 Daltons. Angewandte Chemie International Edition 44, 1504–1508, (2005). - PubMed
-
- Chevillard F & Kolb P SCUBIDOO: a large yet screenable and easily searchable database of computationally created chemical compounds optimized toward high likelihood of synthetic tractability. Journal of chemical information and modeling 55, 1824–1835 (2015). - PubMed
-
- Keserü GM & Makara GM The influence of lead discovery strategies on the properties of drug candidates. Nature Reviews Drug Discovery 8, 203, (2009). - PubMed
References to Online Methods
-
- AMSOL 7.1, University of Minnesota, Minneapolis: (2004).
-
- Wei BQ, Baase WA, Weaver LH, Matthews BW & Shoichet BK A model binding site for testing scoring functions in molecular docking. Journal of molecular biology 322, 339–355 (2002). - PubMed
-
- Mysinger MM & Shoichet BK Rapid context-dependent ligand desolvation in molecular docking. Journal of chemical information and modeling 50, 1561–1573 (2010). - PubMed
