

The Landscape of Circular RNA in Cancer
- PMID: 30735636
- PMCID: PMC6601354
- DOI: 10.1016/j.cell.2018.12.021
The Landscape of Circular RNA in Cancer
Abstract
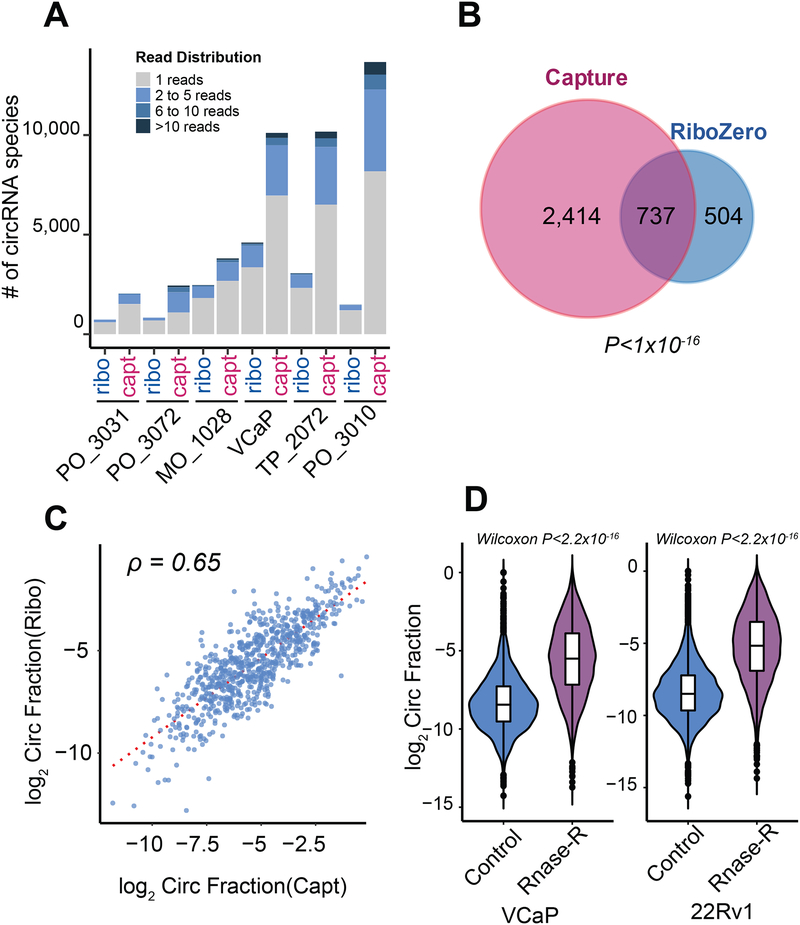
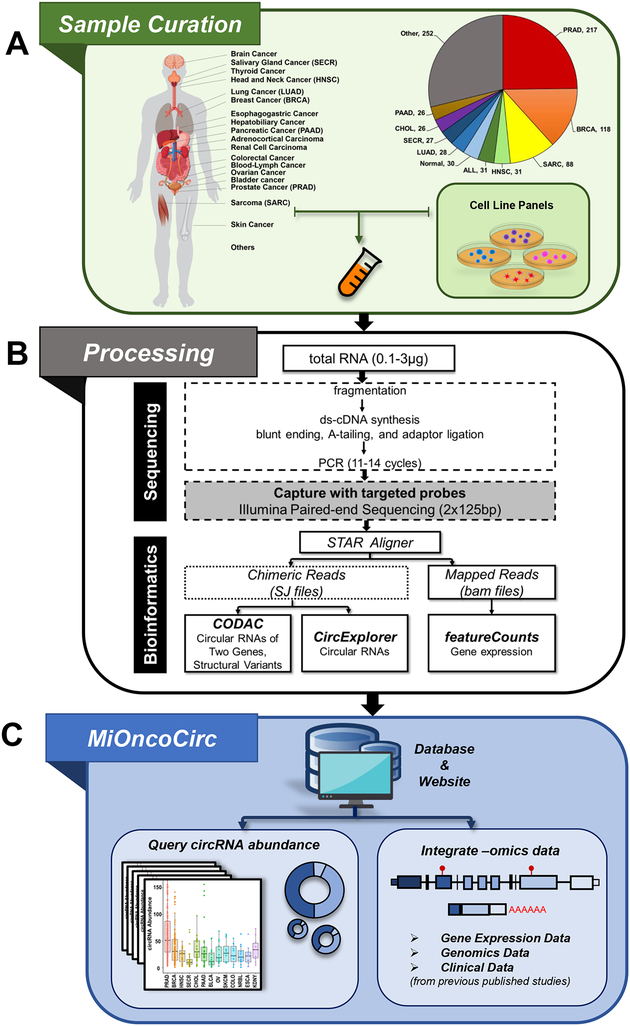
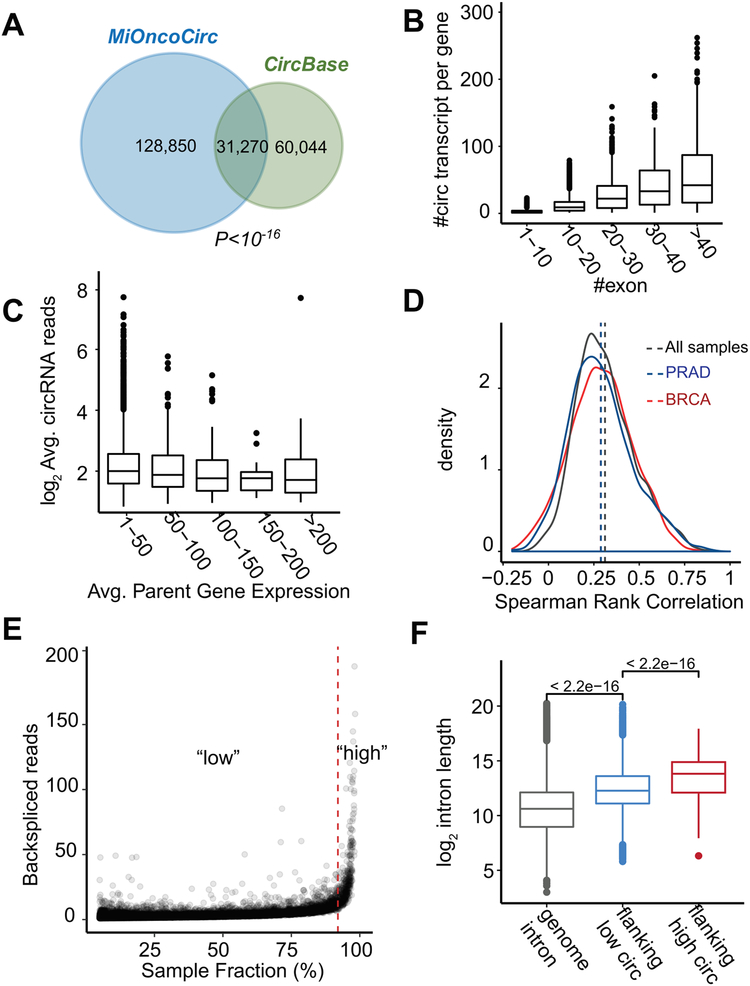
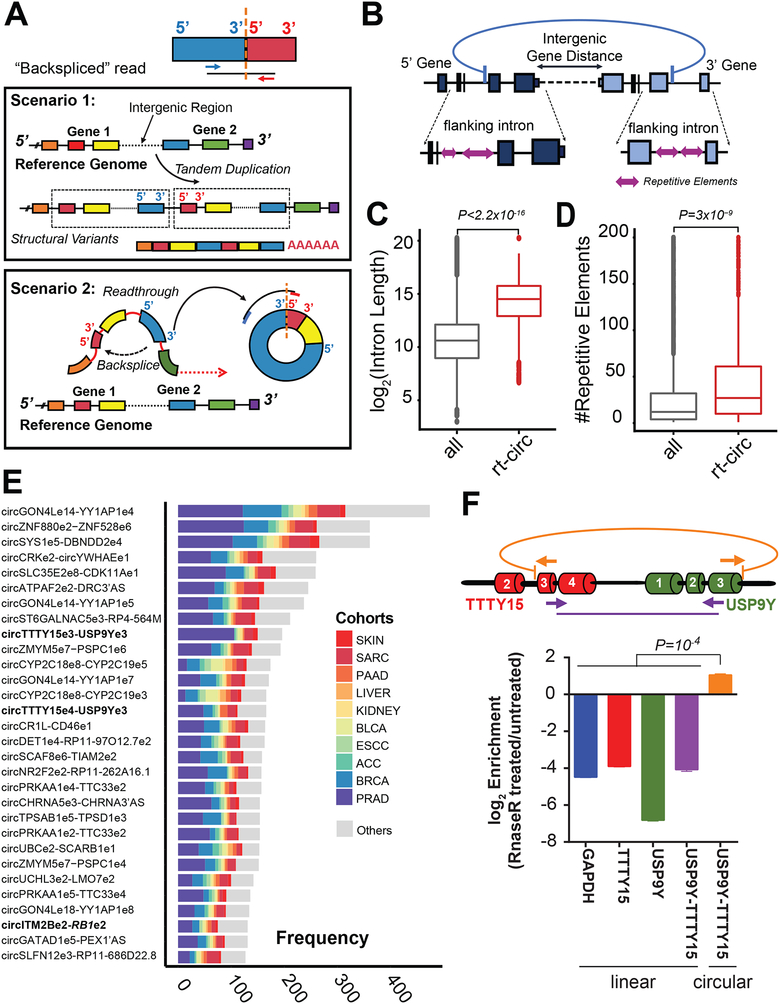
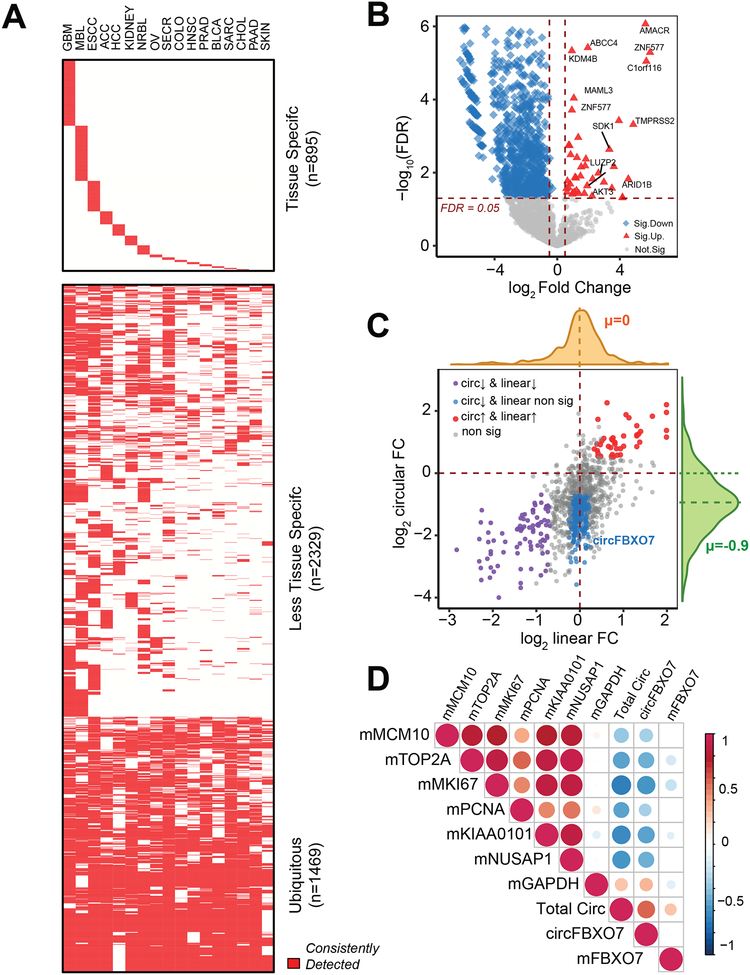
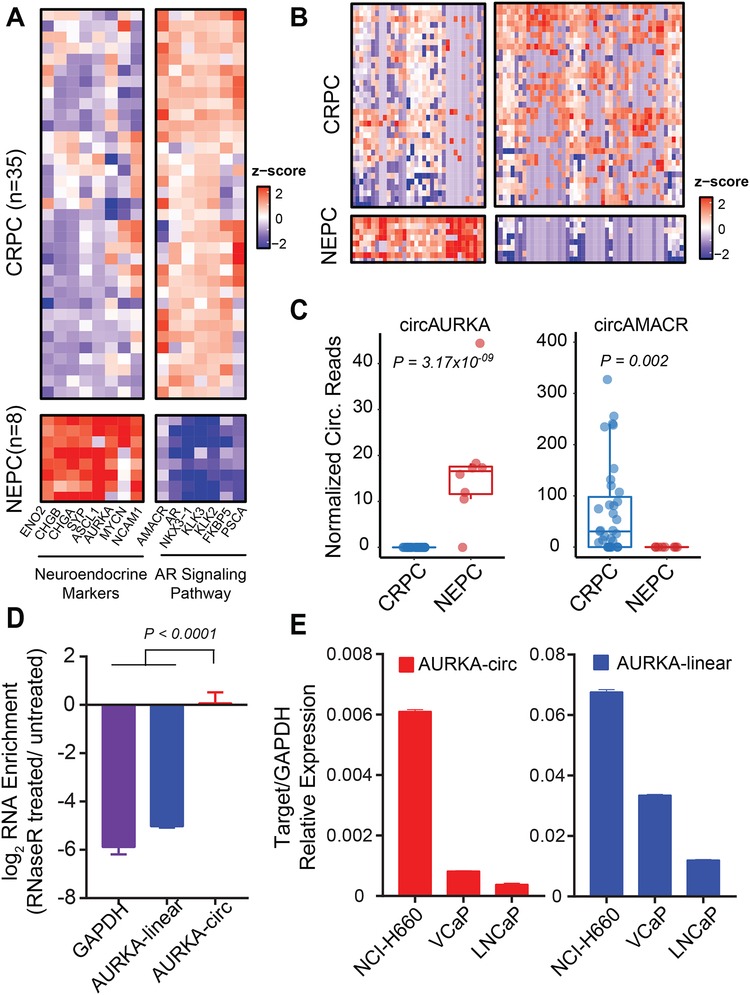
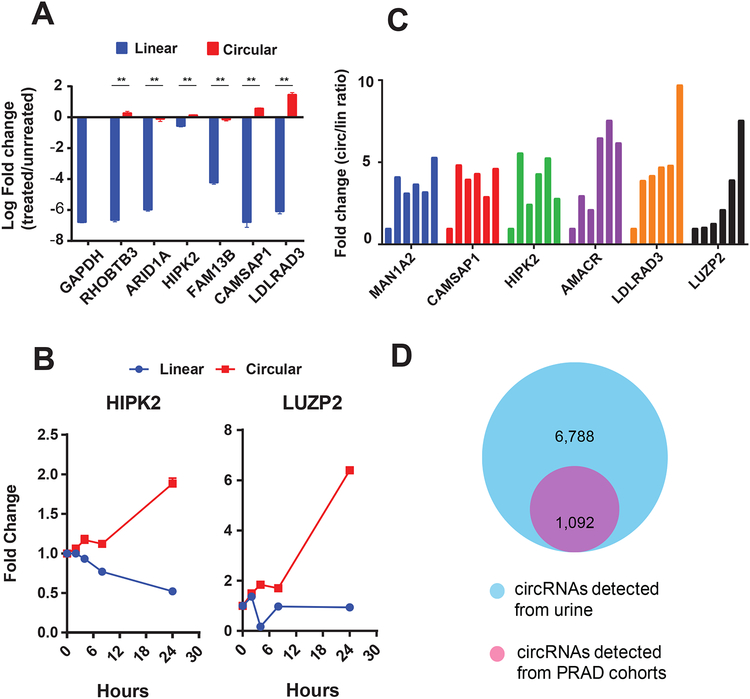
Circular RNAs (circRNAs) are an intriguing class of RNA due to their covalently closed structure, high stability, and implicated roles in gene regulation. Here, we used an exome capture RNA sequencing protocol to detect and characterize circRNAs across >2,000 cancer samples. When compared against Ribo-Zero and RNase R, capture sequencing significantly enhanced the enrichment of circRNAs and preserved accurate circular-to-linear ratios. Using capture sequencing, we built the most comprehensive catalog of circRNA species to date: MiOncoCirc, the first database to be composed primarily of circRNAs directly detected in tumor tissues. Using MiOncoCirc, we identified candidate circRNAs to serve as biomarkers for prostate cancer and were able to detect circRNAs in urine. We further detected a novel class of circular transcripts, termed read-through circRNAs, that involved exons originating from different genes. MiOncoCirc will serve as a valuable resource for the development of circRNAs as diagnostic or therapeutic targets across cancer types.
Keywords: biomarkers; cancer; circRNA; circRNA database; exome capture sequencing; non-coding RNA; read-through transcripts.
Copyright © 2018 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
Comment in
-
Coming full circle in cancer.Nat Rev Genet. 2019 Apr;20(4):191. doi: 10.1038/s41576-019-0104-8. Nat Rev Genet. 2019. PMID: 30804444 No abstract available.
References
-
- Ashwal-Fluss R, Meyer M, Pamudurti N, Ivanov A, Bartok O, Hanan M, Evantal N, Memczak S, Rajewsky N, and Kadener S (2014). circRNA Biogenesis Competes with Pre-mRNA Splicing. Molecular Cell 56, 55–66. - PubMed
-
- Bachmayr-Heyda A, Reiner A, Auer K, Sukhbaatar N, Aust S, Bachleitner-Hofmann T, Mesteri I, Grunt T, Zeillinger R, and Pils D (2015). Correlation of circular RNA abundance with proliferation - exemplified with colorectal and ovarian cancer, idiopathic lung fibrosis and normal human tissues. Scientific Reports 5. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
