

Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease
- PMID: 30737462
- PMCID: PMC9382875
- DOI: 10.1038/s41583-019-0132-6
Oxidative stress, dysfunctional glucose metabolism and Alzheimer disease
Abstract
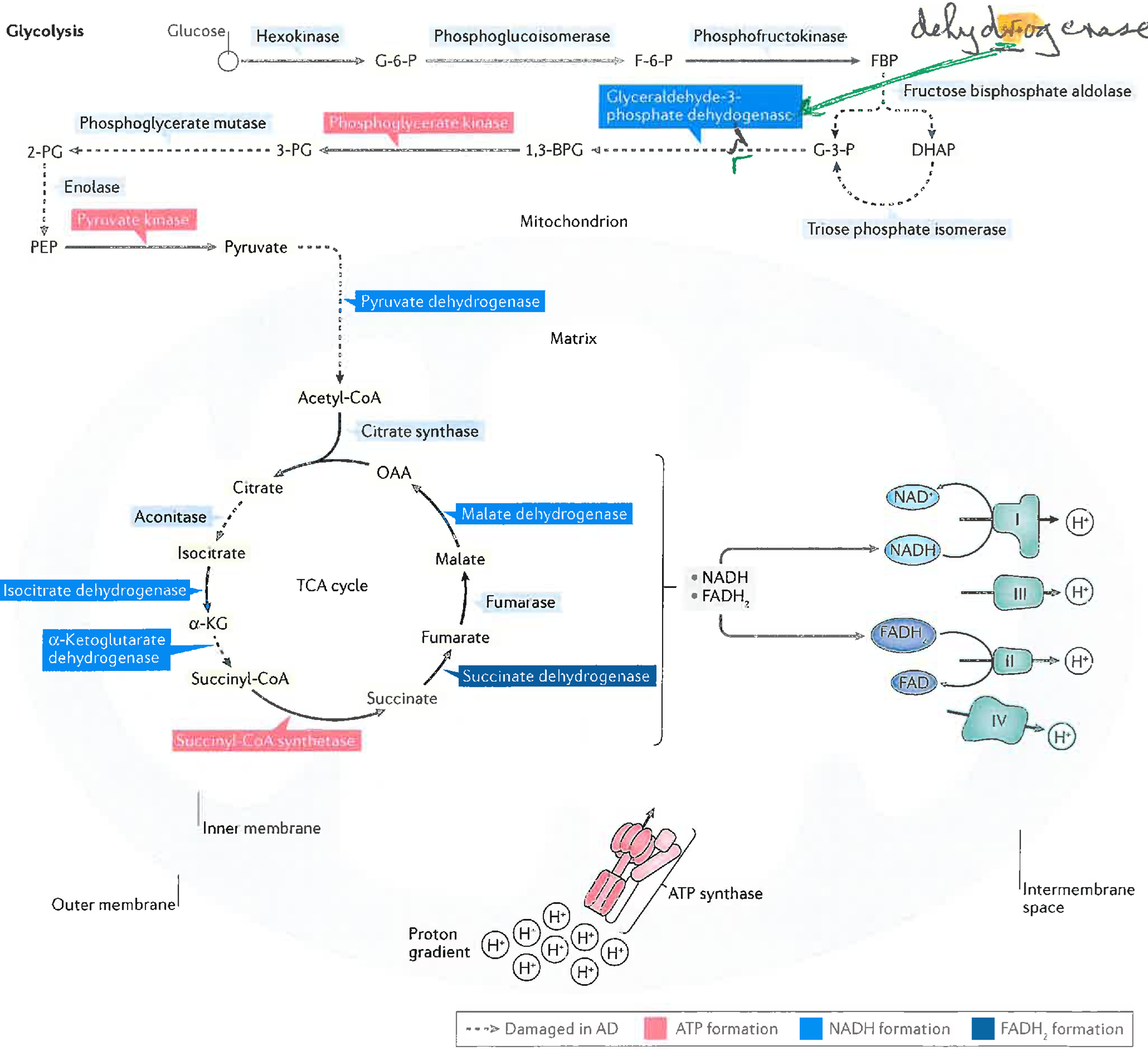
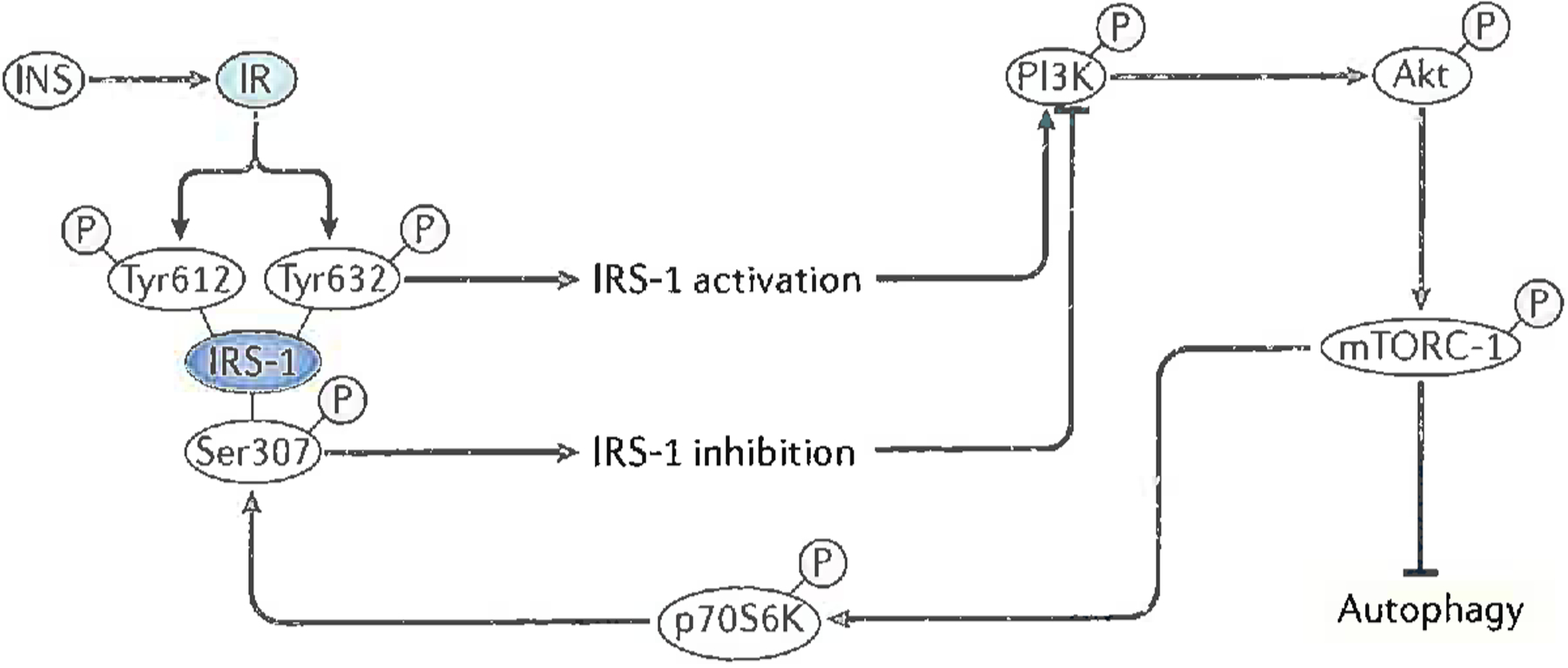
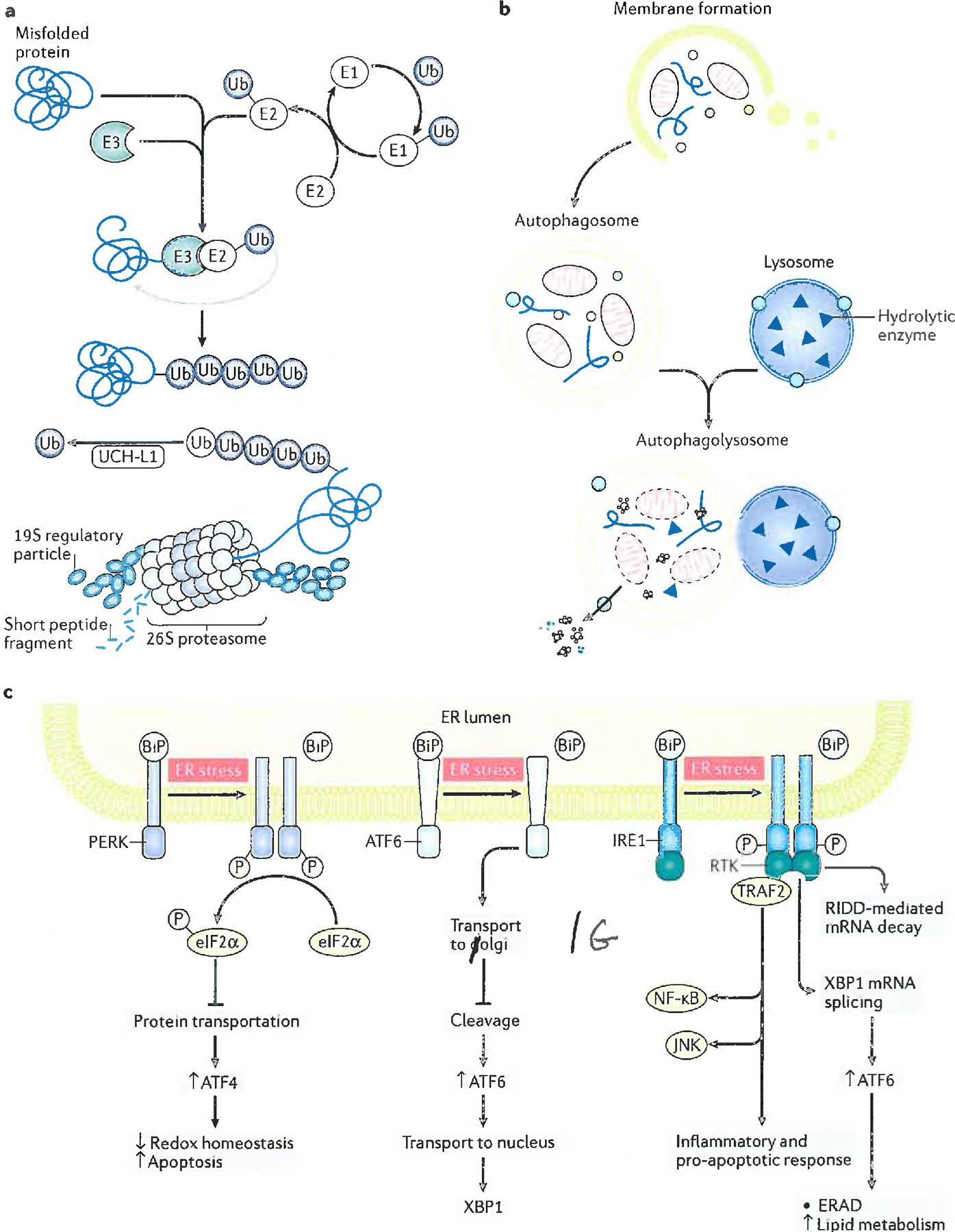
Alzheimer disease (AD) is a major cause of age-related dementia. We do not fully understand AD aetiology and pathogenesis, but oxidative damage is a key component. The brain mostly uses glucose for energy, but in AD and amnestic mild cognitive impairment glucose metabolism is dramatically decreased, probably owing, at least in part, to oxidative damage to enzymes involved in glycolysis, the tricarboxylic acid cycle and ATP biosynthesis. Consequently, ATP-requiring processes for cognitive function are impaired, and synaptic dysfunction and neuronal death result, with ensuing thinning of key brain areas. We summarize current research on the interplay and sequence of these processes and suggest potential pharmacological interventions to retard AD progression.
Conflict of interest statement
Competing interests
Competing interests policy There is NO Competing Interest.
Figures
References
-
- Martins RN et al. Alzheimer’s disease: a journey from amyloid peptides and oxidative stress, to biomarker technologies and disease prevention strategies—gains from AIBL and DIAN cohort studies. J. Alzheimers Dis 62, 965–992 (2018). - PMC - PubMed
-
A detailed review of how cohort studies have contributed to our understanding of AD
-
- Landau SM & Frosch MP Tracking the earliest pathological changes in Alzheimer disease. Neurology 82, 878–883 (2014). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
