

Homogentisate 1,2-dioxygenase (HGD) gene variants, their analysis and genotype-phenotype correlations in the largest cohort of patients with AKU
- PMID: 30737480
- PMCID: PMC6777518
- DOI: 10.1038/s41431-019-0354-0
Homogentisate 1,2-dioxygenase (HGD) gene variants, their analysis and genotype-phenotype correlations in the largest cohort of patients with AKU
Abstract
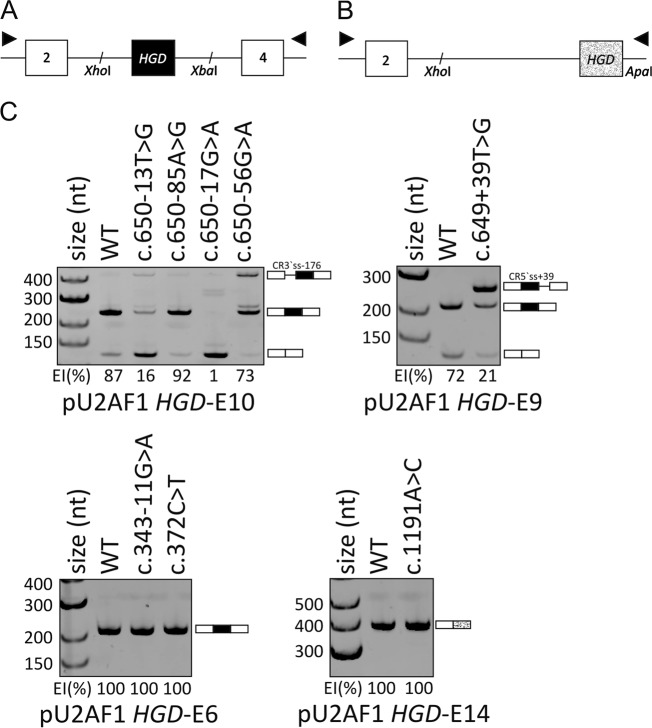
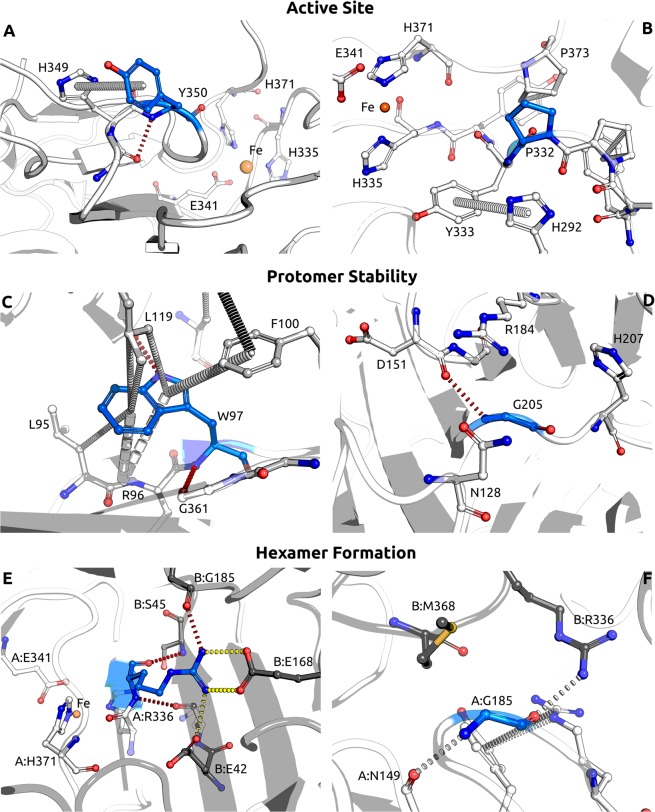
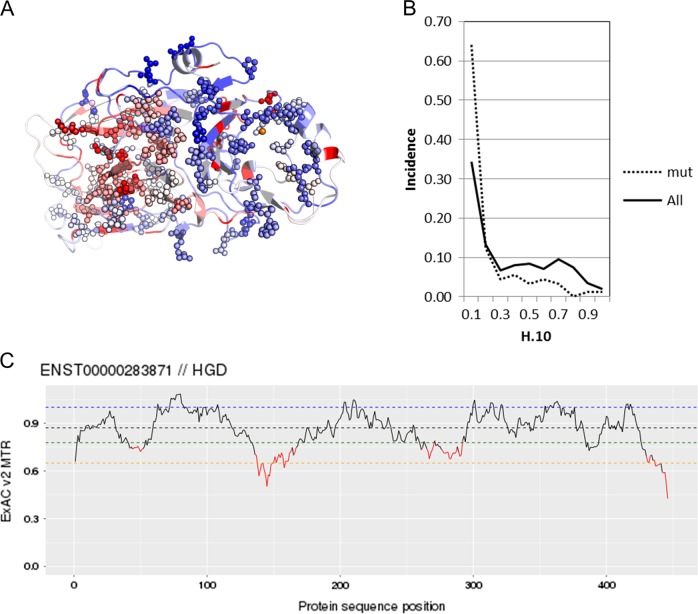
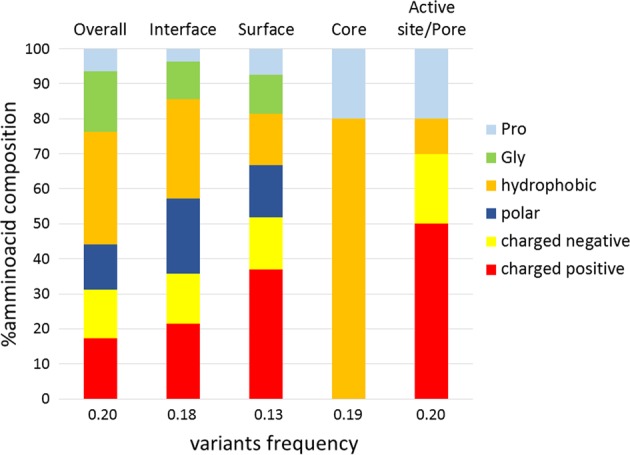
Alkaptonuria (AKU) is a rare metabolic disorder caused by a deficient enzyme in the tyrosine degradation pathway, homogentisate 1,2-dioxygenase (HGD). In 172 AKU patients from 39 countries, we identified 28 novel variants of the HGD gene, which include three larger genomic deletions within this gene discovered via self-designed multiplex ligation-dependent probe amplification (MLPA) probes. In addition, using a reporter minigene assay, we provide evidence that three of eight tested variants potentially affecting splicing cause exon skipping or cryptic splice-site activation. Extensive bioinformatics analysis of novel missense variants, and of the entire HGD monomer, confirmed mCSM as an effective computational tool for evaluating possible enzyme inactivation mechanisms. For the first time for AKU, a genotype-phenotype correlation study was performed for the three most frequent HGD variants identified in the Suitability Of Nitisinone in Alkaptonuria 2 (SONIA2) study. We found a small but statistically significant difference in urinary homogentisic acid (HGA) excretion, corrected for dietary protein intake, between variants leading to 1% or >30% residual HGD activity. There was, interestingly, no difference in serum levels or absolute urinary excretion of HGA, or clinical symptoms, indicating that protein intake is more important than differences in HGD variants for the amounts of HGA that accumulate in the body of AKU patients.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
-
- Garrod AE. Croonian lectures on inborn errors of metabolism, lecture II: alkaptonuria. Lancet. 1908;2:73–79.
-
- La DuBN, Zannoni VG, Laster L, Seegmiller JE. The nature of the defect in tyrosine metabolism in alcaptonuria. J Biol Chem. 1958;230:251–60. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
