

Mitochondrial disorders
- PMID: 30740406
- PMCID: PMC6331360
- DOI: 10.21037/atm.2018.12.13
Mitochondrial disorders
Abstract
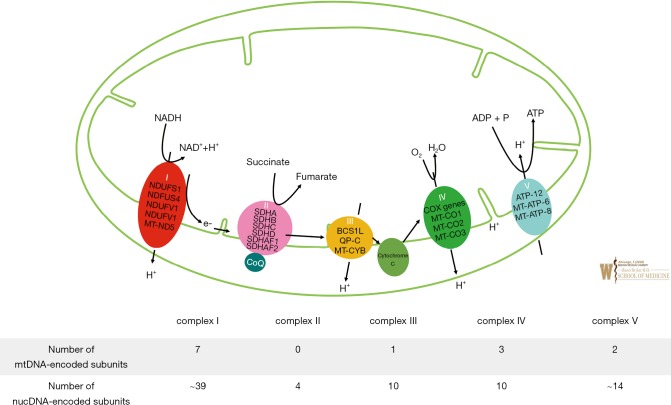
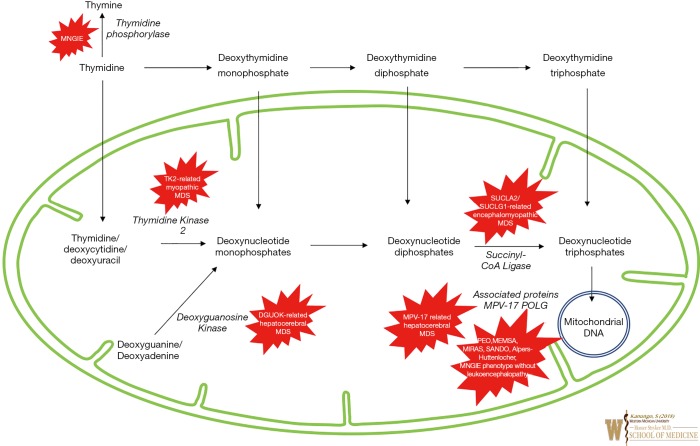
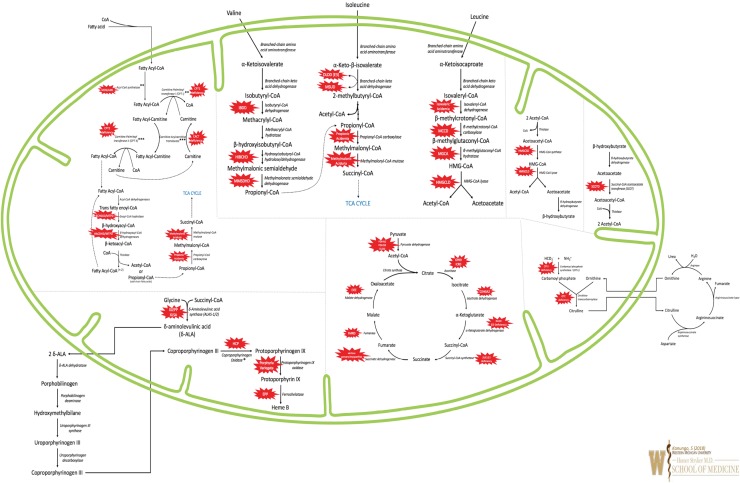
Primary mitochondrial disorders are a group of clinically variable and heterogeneous inborn errors of metabolism (IEMs), resulting from defects in cellular energy, and can affect every organ system of the body. Clinical presentations vary and may include symptoms of fatigue, skeletal muscle weakness, exercise intolerance, short stature, failure to thrive, blindness, ptosis and ophthalmoplegia, nystagmus, hearing loss, hypoglycemia, diabetes mellitus, learning difficulties, intellectual disability, seizures, stroke-like episodes, spasticity, dystonia, hypotonia, pain, neuropsychiatric symptoms, gastrointestinal reflux, dysmotility, gastrointestinal pseudo-obstruction, cardiomyopathy, cardiac conduction defects, and other endocrine, renal, cardiac, and liver problems. Most phenotypic manifestations are multi-systemic, with presentations varying at different age of onset and may show great variability within members of the same family; making these truly complex IEMs. Most primary mitochondrial diseases are autosomal recessive (AR); but maternally-inherited [from mitochondrial (mt) DNA], autosomal dominant and X-linked inheritance are also known. Mitochondria are unique energy-generating cellular organelles, geared for survival and contain their own unique genetic coding material, a circular piece of mtDNA about 16,000 base pairs in size. Additional nuclear (n)DNA encoded genes maintain mitochondrial biogenesis by supervising mtDNA replication, repair and synthesis, which is modified during increased energy demands or physiological stress. Despite our growing knowledge of the hundreds of genetic etiologies for this group of disorders, diagnosis can also remain elusive due to unique aspects of mitochondrial genetics. Though cure and FDA-approved therapies currently elude these IEMs, and current suggested therapies which include nutritional supplements and vitamins are of questionable efficacy; multi-center, international clinical trials are in progress for primary mitochondrial disorders.
Keywords: Mitochondria; energy metabolism; heteroplasmy; mtDNA; nDNA.
Conflict of interest statement
Conflicts of Interest: A Goldstein is an active past president of Mitochondrial Medicine Society (mitosoc.org), Scientific and Medical Advisory Board member of UMDF (umdf.org). The other authors have no conflicts of interest to declare.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Research Materials