

Endocrine and metabolic complications in children and adolescents with Sickle Cell Disease: an Italian cohort study
- PMID: 30744584
- PMCID: PMC6371531
- DOI: 10.1186/s12887-019-1423-9
Endocrine and metabolic complications in children and adolescents with Sickle Cell Disease: an Italian cohort study
Abstract
Background: Children with Sickle Cell Disease (SCD) show endocrine complications and metabolic alterations. The physiopathology of these conditions is not completely understood: iron overload due to chronic transfusions, ischemic damage, and inflammatory state related to vaso-occlusive crises may be involved. Aims of this study were to evaluate the growth pattern, endocrine complications, and metabolic alterations and to detect the relationship between these conditions and the SCD severity in affected children and adolescents.
Methods: Fifty-two children and adolescents with SCD [38 homozygous sickle hemoglobin (HbSS) and 14 heterozygous sickle hemoglobin (HbSC); age range 3-18 years] were recruited. Anthropometric [height, body mass index (BMI), arm span, sitting height, target height (TH), and pubertal status] and laboratory [blood cell counts, hemolysis indices, metabolic and nutritional status indices and hormonal blood levels] data were evaluated. The SCD severity was defined according to hematological and clinical parameters.
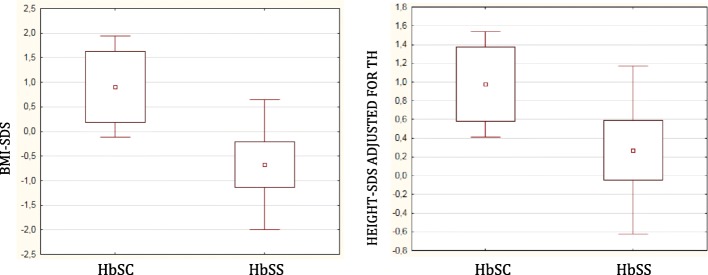
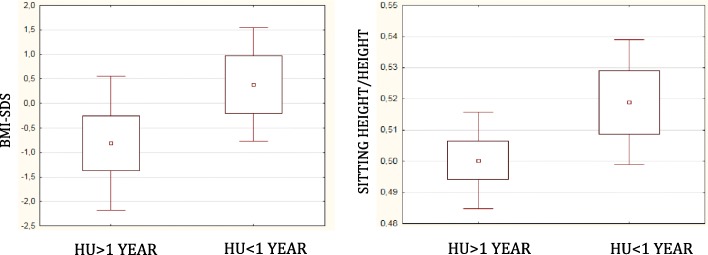
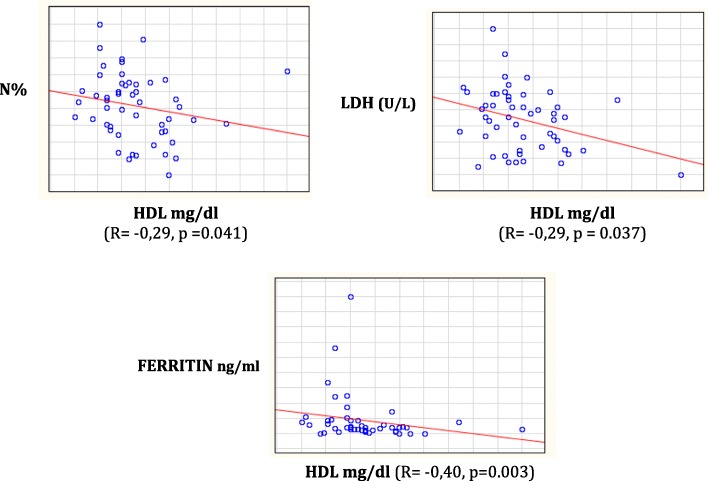
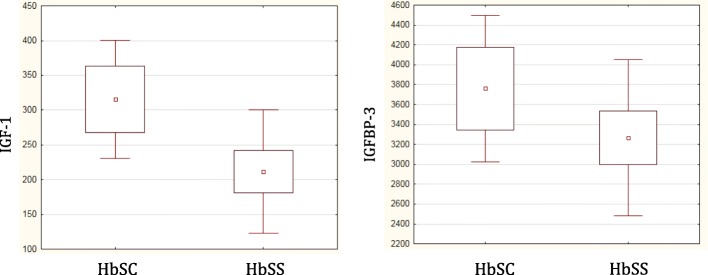
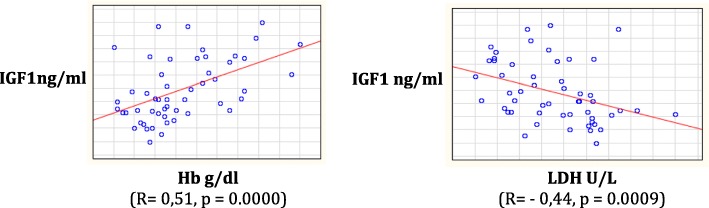
Results: Height-SDS adjusted for TH and BMI-SDS were significantly higher in HbSC children than in HbSS ones. Forty-eight out of 52 patients (92%) had at least one metabolic and/or endocrine alteration: insufficiency/deficiency of vitamin D (84.7%), insulin resistance (11.5%), growth hormone deficiency (3.8%), subclinical hypothyroidism (3.8%), and hypogonadism (1.9%). Levels of vitamin D were significantly and negatively correlated with clinical indicators of the SCD severity. Subjects with HbSS genotype show significant lower levels of both insulin-like growth factor-1 (IGF-1) and insulin-like growth factor binding protein 3 than children with HbSC. In the study population IGF-1 values were significantly and positively correlated with Hb and negatively with lactate dehydrogenase.
Conclusions: Metabolic alterations and endocrine complications are very common in children and adolescents with SCD. A regular follow-up is necessary to identify subjects at risk for complications to precociously start an appropriate treatment and to improve the quality of life of SCD patients.
Keywords: Children and adolescents; Endocrine complications; Metabolism; Sickle cell disease.
Conflict of interest statement
Ethics approval and consent to participate
Provincial Ethical Committee approved the protocol study (E.C. n. 213/16), informed consent was obtained for all enrolled patients. Parents and/or legal guardians provided the written informed consent for participation on behalf of the underage participants who were not of legal age to consent for themselves.
Consent for publication
Not applicable
Competing interests
Lorenzo Iughetti is an Editorial Board Member for BMC Pediatrics. All the others authors declared that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
