

Identification of potential blood biomarkers for Parkinson's disease by gene expression and DNA methylation data integration analysis
- PMID: 30744671
- PMCID: PMC6371578
- DOI: 10.1186/s13148-019-0621-5
Identification of potential blood biomarkers for Parkinson's disease by gene expression and DNA methylation data integration analysis
Abstract
Background: Blood-based gene expression or epigenetic biomarkers of Parkinson's disease (PD) are highly desirable. However, accuracy and specificity need to be improved, and methods for the integration of gene expression with epigenetic data need to be developed in order to make this feasible.
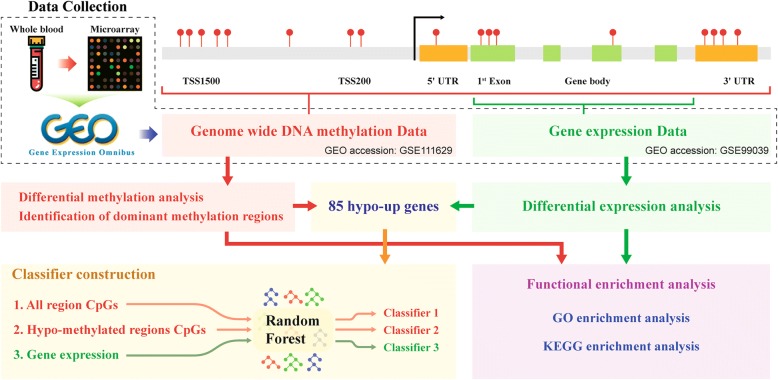
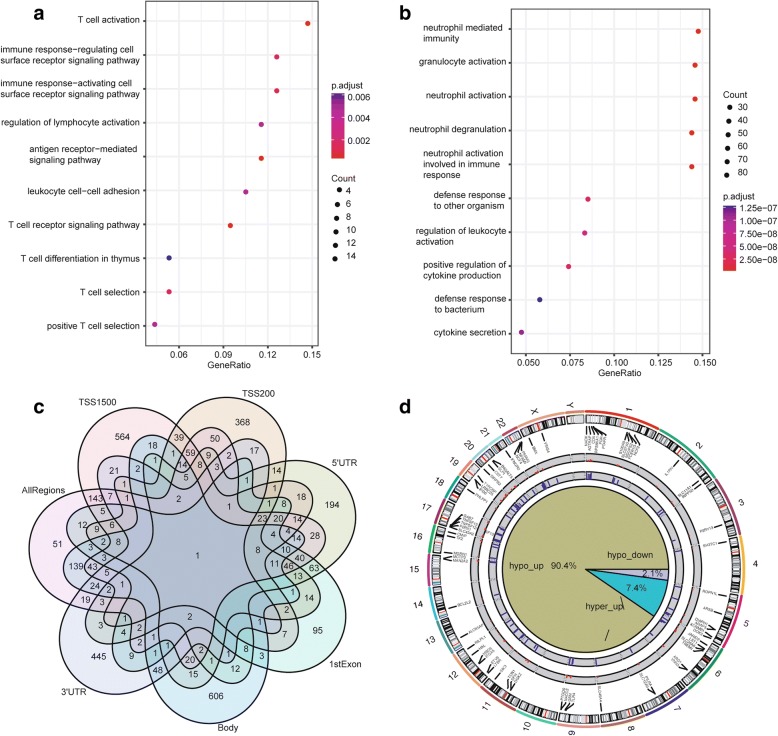
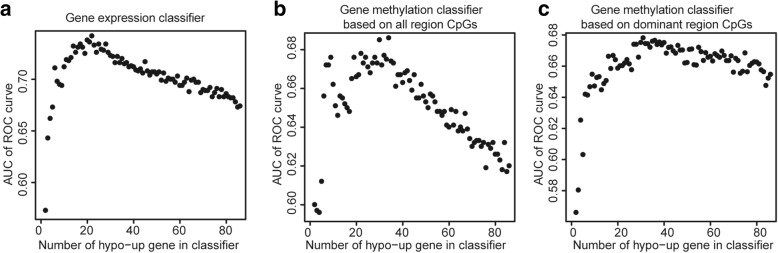
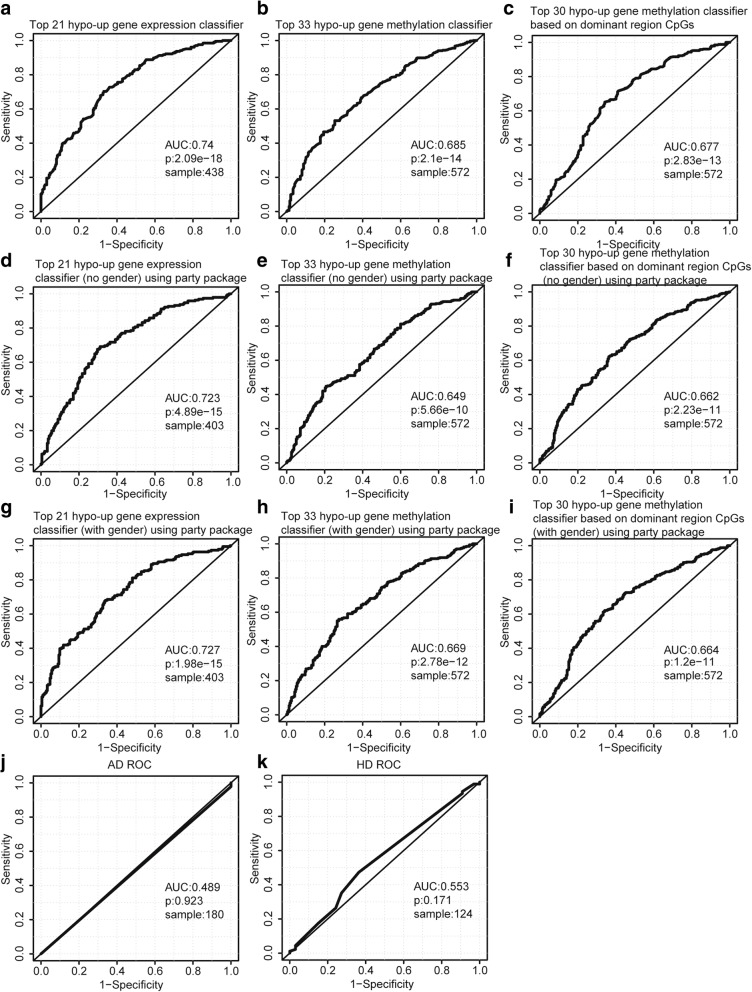
Methods: Whole blood gene expression data and DNA methylation data were downloaded from Gene Expression Omnibus (GEO) database. A linear model was used to identify significantly differentially expressed genes (DEGs) and differentially methylated genes (DMGs) according to specific gene regions 5'-C-phosphate-G-3' (CpGs) or all gene regions CpGs in PD. Gene set enrichment analysis was then applied to DEGs and DMGs. Subsequently, data integration analysis was performed to identify robust PD-associated blood biomarkers. Finally, the random forest algorithm and a leave-one-out cross validation method were performed to construct classifiers based on gene expression data integrated with methylation data.
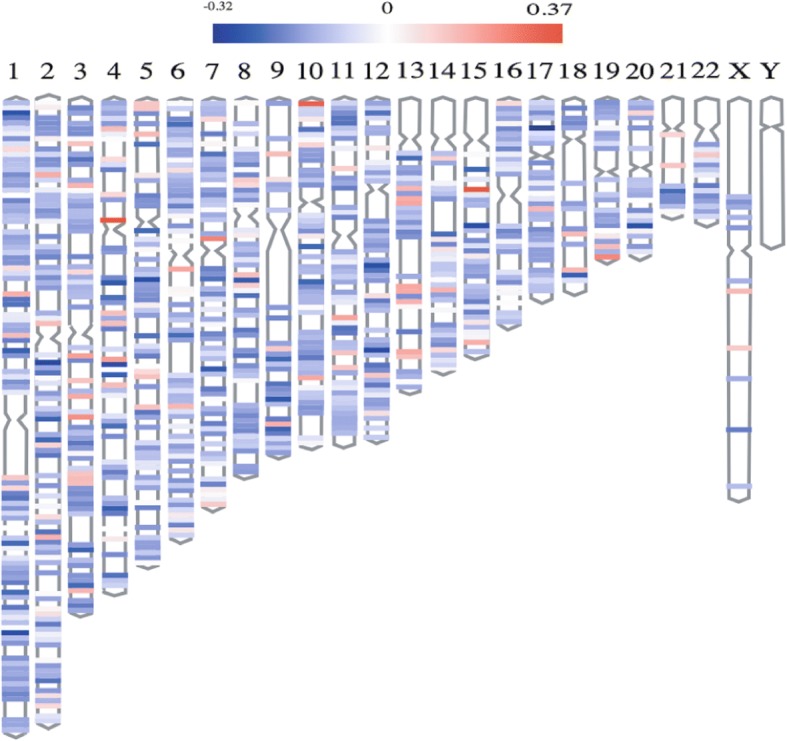
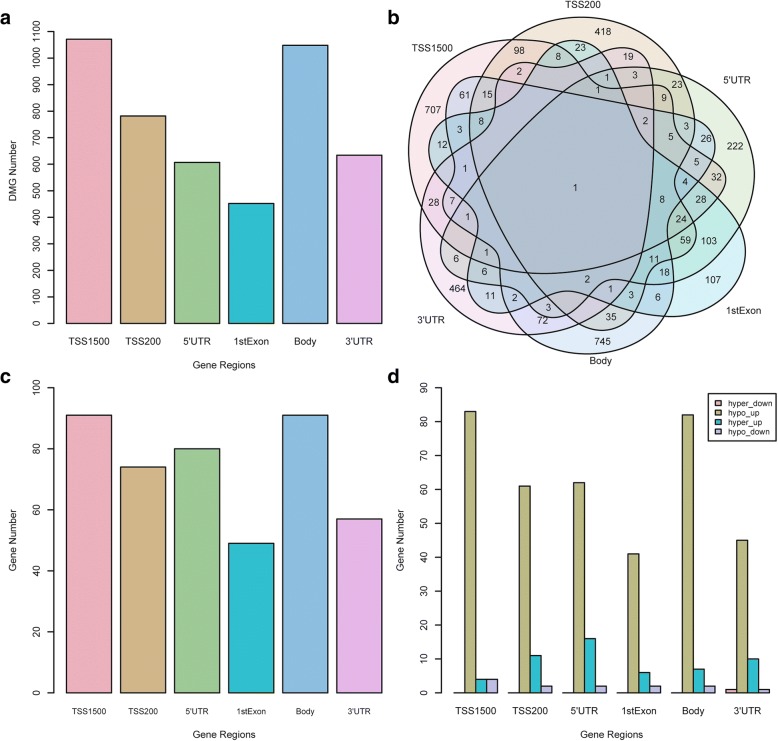
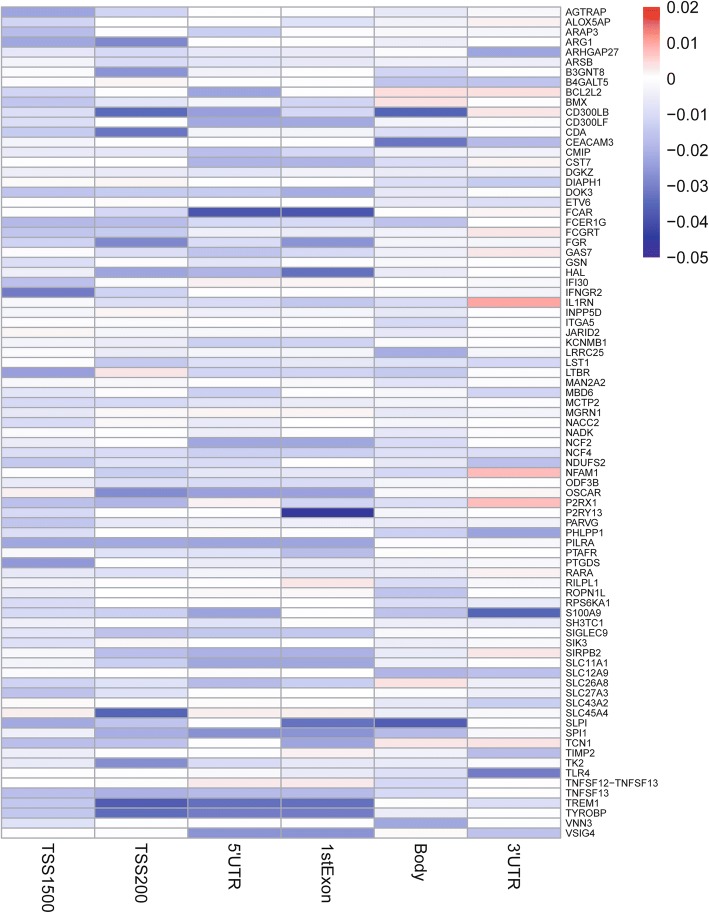
Results: Eighty-five (85) significantly hypo-methylated and upregulated genes in PD patients compared to healthy controls were identified. The dominant hypo-methylated regions of these genes were significantly different. Some genes had a single dominant hypo-methylated region, while others had multiple dominant hypo-methylated regions. One gene expression classifier and two gene methylation classifiers based on all or dominant methylation-altered region CpGs were constructed. All have a good prediction power for PD.
Conclusions: Gene expression and methylation data integration analysis identified a blood-based 53-gene signature, which could be applied as a biomarker for PD.
Keywords: DNA methylation; Data integration; Gene expression; Parkinson’s disease.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
