

CellFishing.jl: an ultrafast and scalable cell search method for single-cell RNA sequencing
- PMID: 30744683
- PMCID: PMC6371477
- DOI: 10.1186/s13059-019-1639-x
CellFishing.jl: an ultrafast and scalable cell search method for single-cell RNA sequencing
Abstract
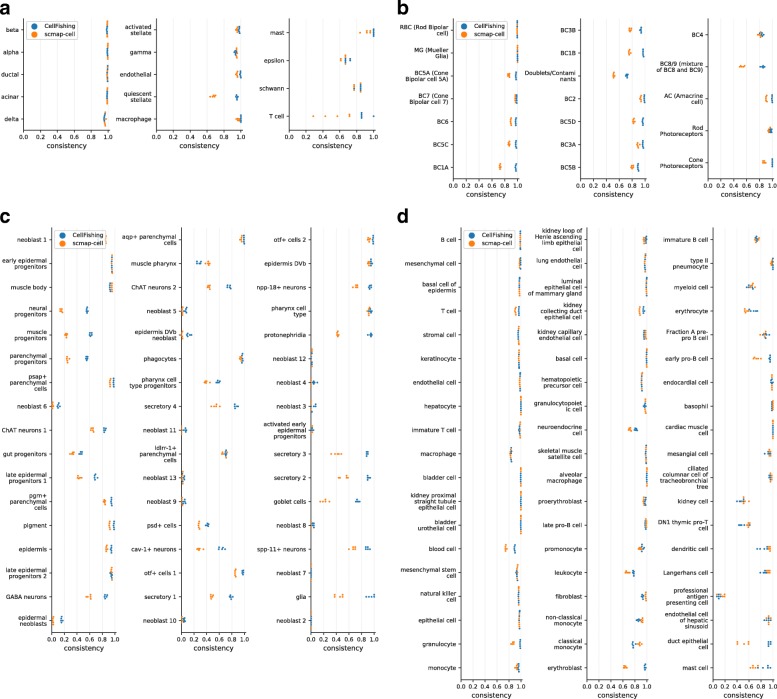
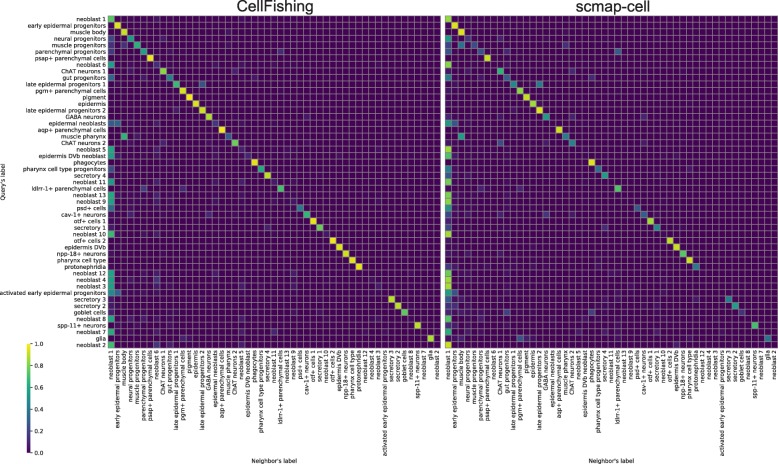
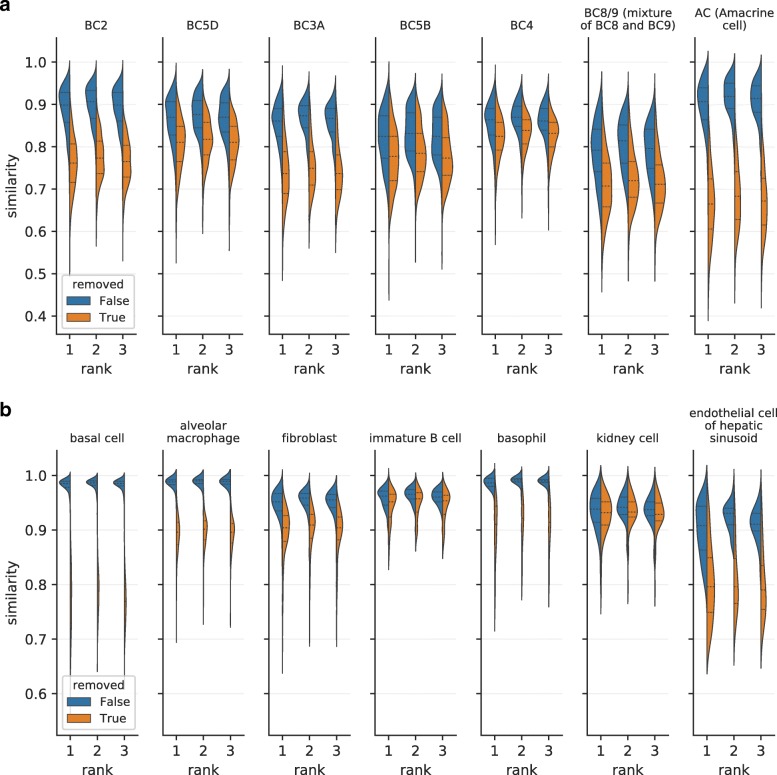
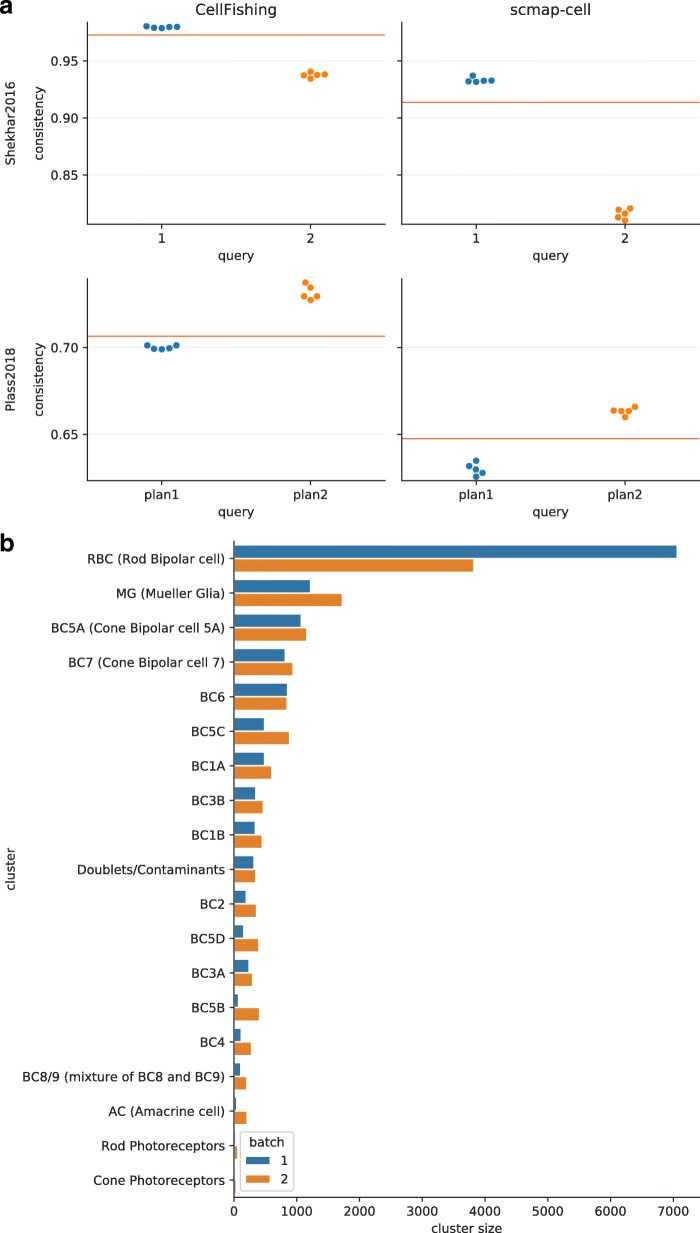
Recent technical improvements in single-cell RNA sequencing (scRNA-seq) have enabled massively parallel profiling of transcriptomes, thereby promoting large-scale studies encompassing a wide range of cell types of multicellular organisms. With this background, we propose CellFishing.jl, a new method for searching atlas-scale datasets for similar cells and detecting noteworthy genes of query cells with high accuracy and throughput. Using multiple scRNA-seq datasets, we validate that our method demonstrates comparable accuracy to and is markedly faster than the state-of-the-art software. Moreover, CellFishing.jl is scalable to more than one million cells, and the throughput of the search is approximately 1600 cells per second.
Keywords: Cell searching; Cell typing; Locality-sensitive hashing; scRNA-seq.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
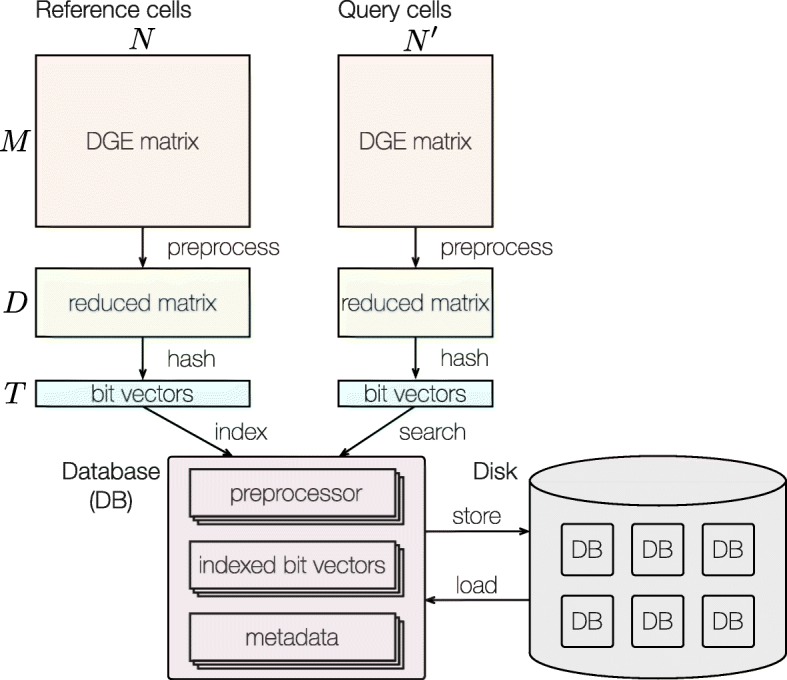
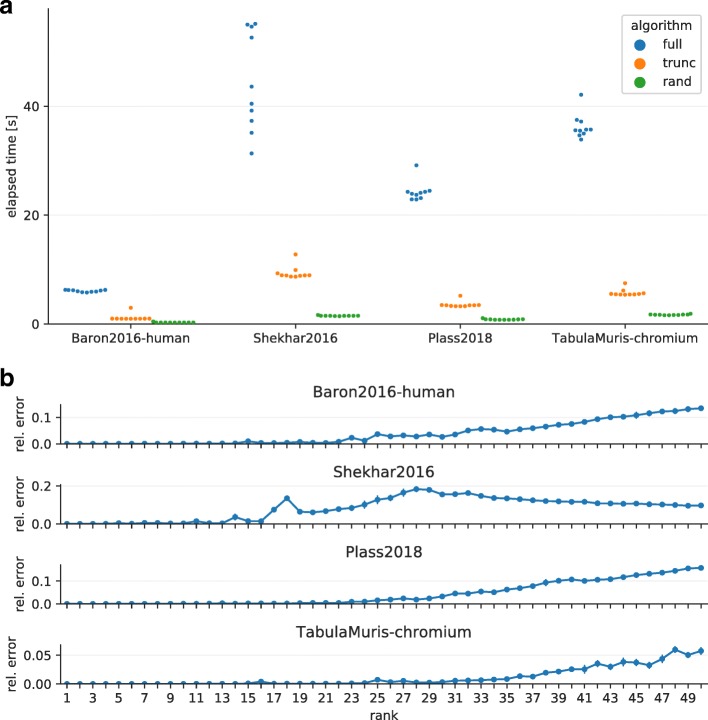
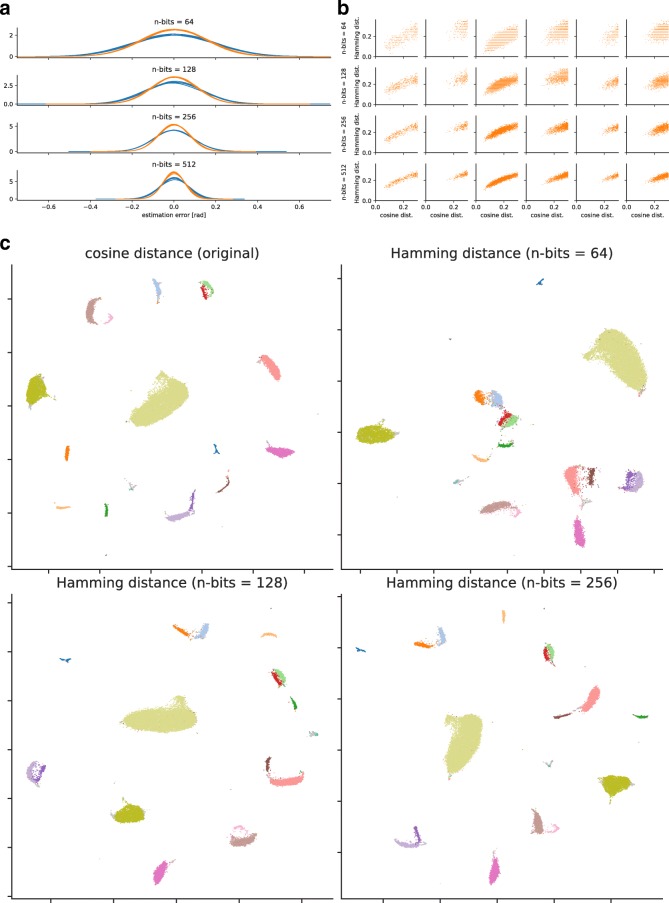
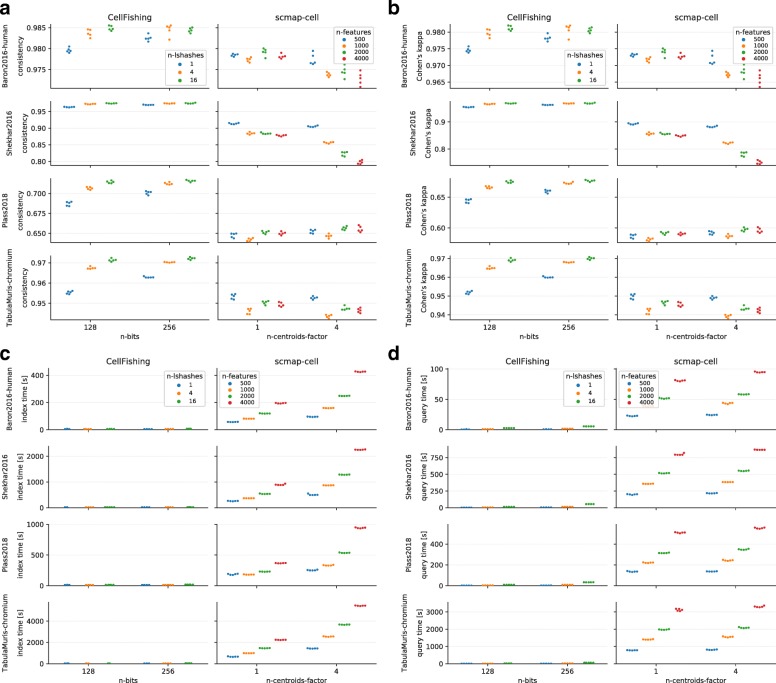
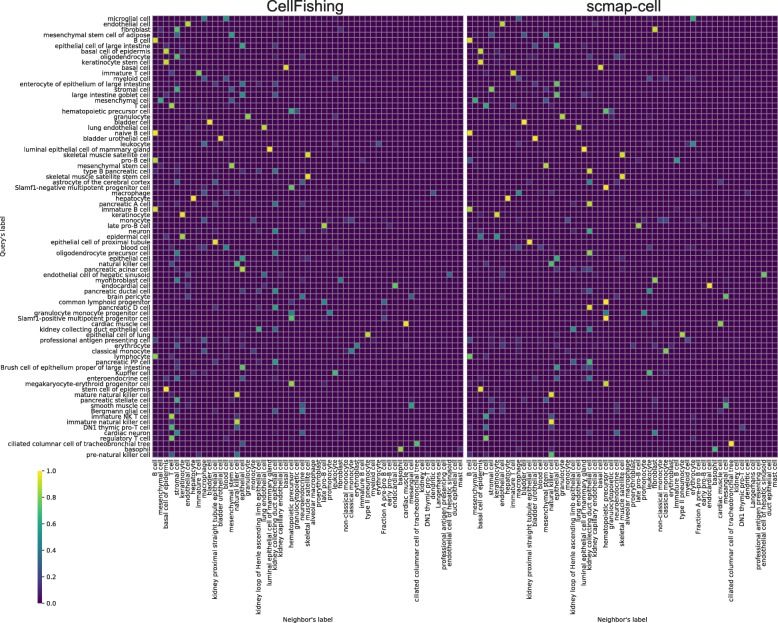
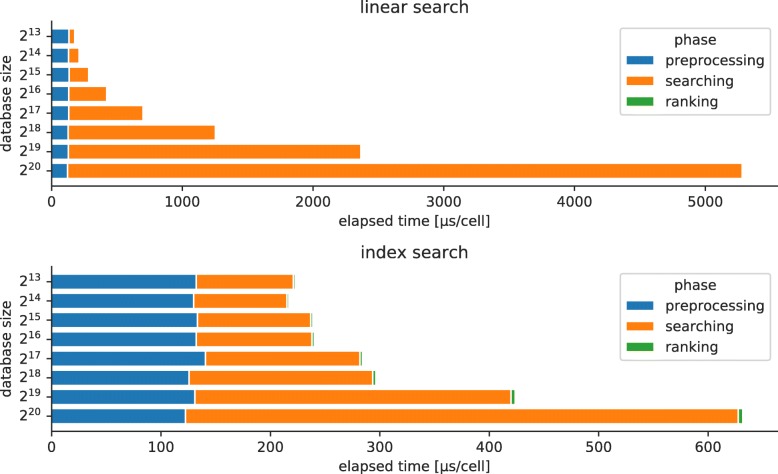
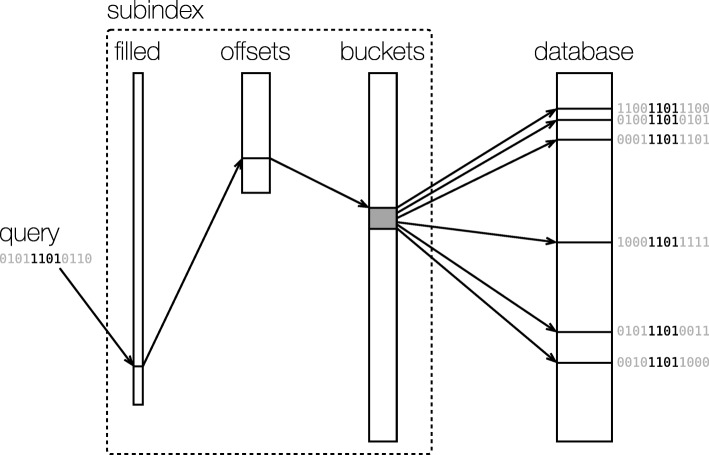
References
-
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: Single-Cell RNA-Seq by Multiplexed Linear Amplification. Cell Rep. 2012; 2(3):666–73. 10.1016/j.celrep.2012.08.003. - PubMed
-
- Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods. 2012; 9(1):72–4. 10.1038/nmeth.1778. - PubMed
-
- Islam S, Zeisel A, Joost S, La Manno G, Zajac P, Kasper M, Lönnerberg P, Linnarsson S. Quantitative single-cell RNA-seq with unique molecular identifiers. Nat Methods. 2014; 11(2):163–6. 10.1038/nmeth.2772. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
