

Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases
- PMID: 30745444
- PMCID: PMC6382364
- DOI: 10.1212/WNL.0000000000006926
Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases
Abstract
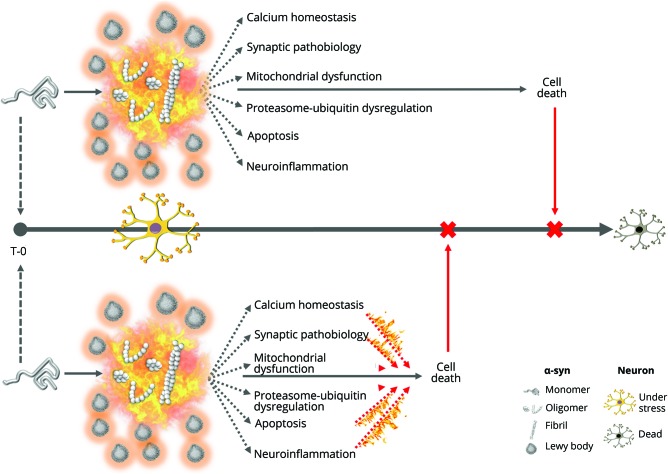
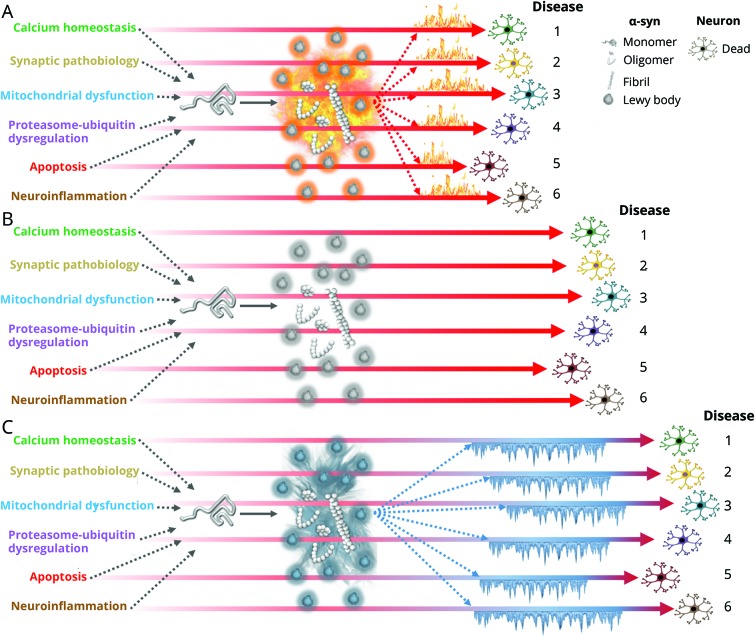
The gold standard for a definitive diagnosis of Parkinson disease (PD) is the pathologic finding of aggregated α-synuclein into Lewy bodies and for Alzheimer disease (AD) aggregated amyloid into plaques and hyperphosphorylated tau into tangles. Implicit in this clinicopathologic-based nosology is the assumption that pathologic protein aggregation at autopsy reflects pathogenesis at disease onset. While these aggregates may in exceptional cases be on a causal pathway in humans (e.g., aggregated α-synuclein in SNCA gene multiplication or aggregated β-amyloid in APP mutations), their near universality at postmortem in sporadic PD and AD suggests they may alternatively represent common outcomes from upstream mechanisms or compensatory responses to cellular stress in order to delay cell death. These 3 conceptual frameworks of protein aggregation (pathogenic, epiphenomenon, protective) are difficult to resolve because of the inability to probe brain tissue in real time. Whereas animal models, in which neither PD nor AD occur in natural states, consistently support a pathogenic role of protein aggregation, indirect evidence from human studies does not. We hypothesize that (1) current biomarkers of protein aggregates may be relevant to common pathology but not to subgroup pathogenesis and (2) disease-modifying treatments targeting oligomers or fibrils might be futile or deleterious because these proteins are epiphenomena or protective in the human brain under molecular stress. Future precision medicine efforts for molecular targeting of neurodegenerative diseases may require analyses not anchored on current clinicopathologic criteria but instead on biological signals generated from large deeply phenotyped aging populations or from smaller but well-defined genetic-molecular cohorts.
© 2019 American Academy of Neurology.
Figures
Comment in
-
Reader response: Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases.Neurology. 2020 Jan 21;94(3):143-144. doi: 10.1212/WNL.0000000000008821. Neurology. 2020. PMID: 31959684 No abstract available.
-
Author response: Revisiting protein aggregation as pathogenic in sporadic Parkinson and Alzheimer diseases.Neurology. 2020 Jan 21;94(3):144-145. doi: 10.1212/WNL.0000000000008822. Neurology. 2020. PMID: 31959685 No abstract available.
References
-
- Chartier-Harlin M-C, Kachergus J, Roumier C, et al. α-Synuclein locus duplication as a cause of familial Parkinson's disease. Lancet 2004;364:1167–1169. - PubMed
-
- Sleegers K, Brouwers N, Gijselinck I, et al. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain 2006;129:2977–2983. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous