

Reliable and practical computational description of molecular crystal polymorphs
- PMID: 30746448
- PMCID: PMC6357866
- DOI: 10.1126/sciadv.aau3338
Reliable and practical computational description of molecular crystal polymorphs
Abstract
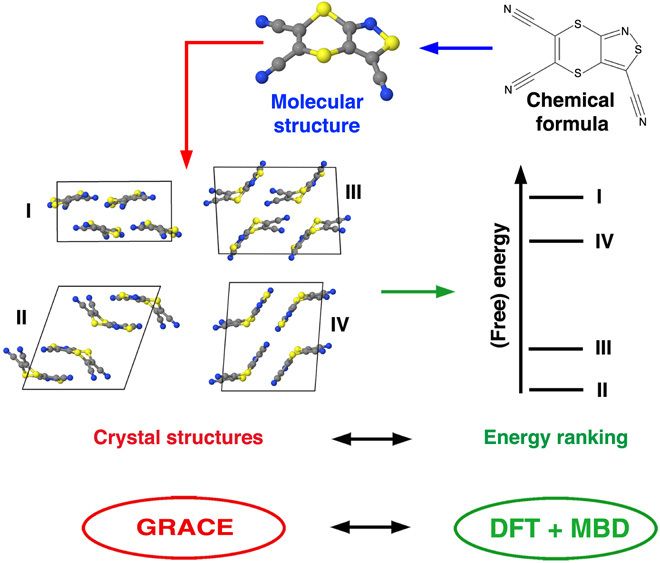
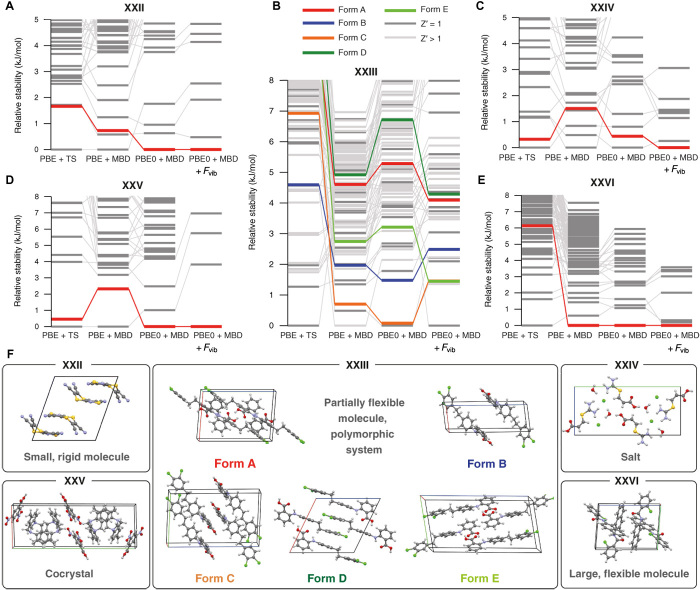
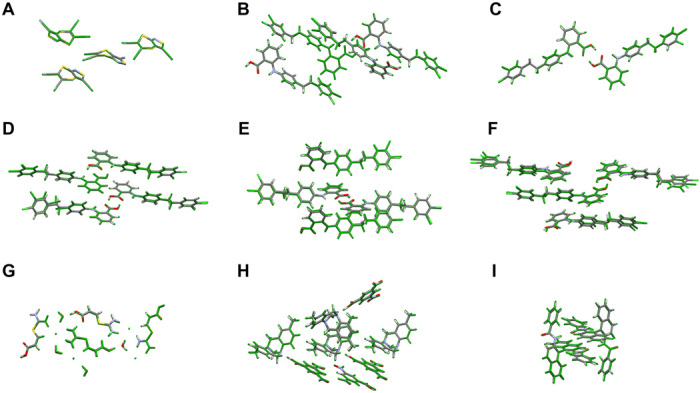
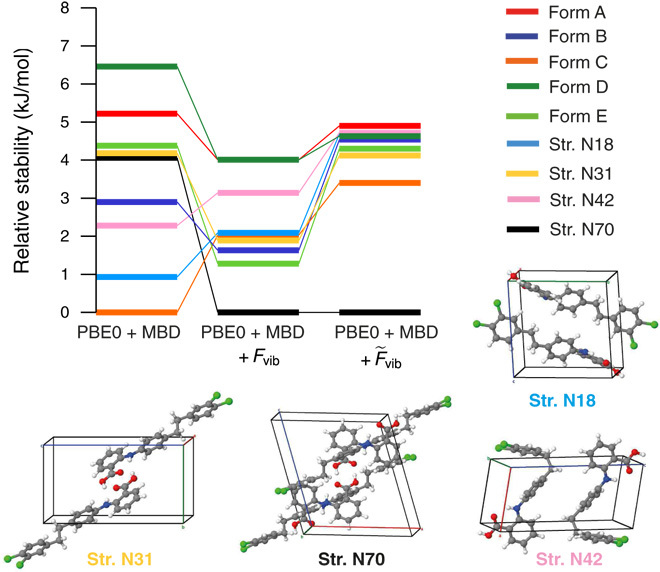
Reliable prediction of the polymorphic energy landscape of a molecular crystal would yield profound insight into drug development in terms of the existence and likelihood of late-appearing polymorphs. However, the computational prediction of molecular crystal polymorphs is highly challenging due to the high dimensionality of conformational and crystallographic space accompanied by the need for relative free energies to within 1 kJ/mol per molecule. In this study, we combine the most successful crystal structure sampling strategy with the most successful first-principles energy ranking strategy of the latest blind test of organic crystal structure prediction methods. Specifically, we present a hierarchical energy ranking approach intended for the refinement of relative stabilities in the final stage of a crystal structure prediction procedure. Such a combined approach provides excellent stability rankings for all studied systems and can be applied to molecular crystals of pharmaceutical importance.
Figures
References
-
- Cruz-Cabeza A. J., Reutzel-Edens S. M., Bernstein J., Facts and fictions about polymorphism. Chem. Soc. Rev. 44, 8619–8635 (2015). - PubMed
-
- Price S. L., Predicting crystal structures of organic compounds. Chem. Soc. Rev. 43, 2098–2111 (2014). - PubMed
-
- Reilly A. M., Cooper R. I., Adjiman C. S., Bhattacharya S., Boese A. D., Brandenburg J. G., Bygrave P. J., Bylsma R., Campbell J. E., Car R., Case D. H., Chadha R., Cole J. C., Cosburn K., Cuppen H. M., Curtis F., Day G. M., DiStasio R. A. Jr., Dzyabchenko A., van Eijck B. P., Elking D. M., van den Ende J. A., Facelli J. C., Ferraro M. B., Fusti-Molnar L., Gatsiou C.-A., Gee T. S., de Gelder R., Ghiringhelli L. M., Goto H., Grimme S., Guo R., Hofmann D. W. M., Hoja J., Hylton R. K., Iuzzolino L., Jankiewicz W., de Jong D. T., Kendrick J., de Klerk N. J. J., Ko H.-Y., Kuleshova L. N., Li X., Lohani S., Leusen F. J. J., Lund A. M., Lv J., Ma Y., Marom N., Masunov A. E., McCabe P., McMahon D. P., Meekes H., Metz M. P., Misquitta A. J., Mohamed S., Monserrat B., Needs R. J., Neumann M. A., Nyman J., Obata S., Oberhofer H., Oganov A. R., Orendt A. M., Pagola G. I., Pantelides C. C., Pickard C. J., Podeszwa R., Price L. S., Price S. L., Pulido A., Read M. G., Reuter K., Schneider E., Schober C., Shields G. P., Singh P., Sugden I. J., Szalewicz K., Taylor C. R., Tkatchenko A., Tuckerman M. E., Vacarro F., Vasileiadis M., Vazquez-Mayagoitia A., Vogt L., Wang Y., Watson R. E., de Wijs G. A., Yang J., Zhu Q., Groom C. R., Report on the sixth blind test of organic crystal structure prediction methods. Acta Crystallogr. B Struct. Sci. Cryst. Eng. Mater. 72, 439–459 (2016). - PMC - PubMed
-
- Beran G. J. O., Modeling polymorphic molecular crystals with electronic structure theory. Chem. Rev. 116, 5567–5613 (2016). - PubMed
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
