

Mitochondrial dysfunctions in barth syndrome
- PMID: 30746873
- PMCID: PMC6586490
- DOI: 10.1002/iub.2018
Mitochondrial dysfunctions in barth syndrome
Abstract
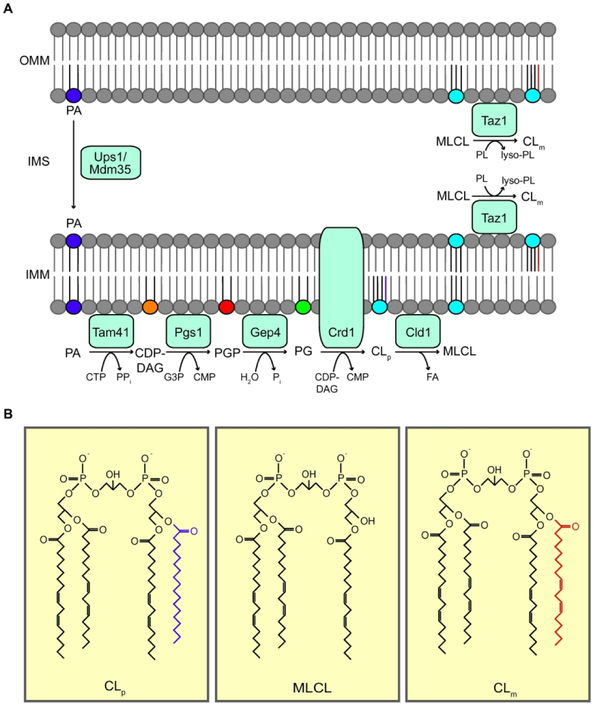
Barth syndrome (BTHS) is a rare multisystemic genetic disorder caused by mutations in the TAZ gene. TAZ encodes a mitochondrial enzyme that remodels the acyl chain composition of newly synthesized cardiolipin, a phospholipid unique to mitochondrial membranes. The clinical abnormalities observed in BTHS patients are caused by perturbations in various mitochondrial functions that rely on remodeled cardiolipin. However, the contribution of different cardiolipin-dependent mitochondrial functions to the pathology of BTHS is not fully understood. In this review, we will discuss recent findings from different genetic models of BTHS, including the yeast model of cardiolipin deficiency that has uncovered the specific in vivo roles of cardiolipin in mitochondrial respiratory chain biogenesis, bioenergetics, intermediary metabolism, mitochondrial dynamics, and quality control. We will also describe findings from higher eukaryotic models of BTHS that highlight a link between cardiolipin-dependent mitochondrial function and its impact on tissue and organ function. © 2019 IUBMB Life, 9999(9999):1-11, 2019.
Keywords: Barth syndrome; cardiolipin; mitochondria.
© 2019 International Union of Biochemistry and Molecular Biology.
Figures
References
-
- Barth PG, Scholte HR, Berden JA, Van der Klei-Van Moorsel JM, Luyt-Houwen IE, et al. (1983) An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes. J. Neurol. Sci. 62, 327–355. - PubMed
-
- Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA, et al. (1996) A novel X-linked gene, G4.5. is responsible for Barth syndrome. Nat. Genet. 12, 385–389. - PubMed
-
- Xu Y, Malhotra A, Ren M, and Schlame M (2006) The enzymatic function of tafazzin. J. Biol. Chem. 281, 39217–39224. - PubMed
-
- Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, et al. (2002) Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome. Ann. Neurol. 51, 634–637. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
