

HDAC inhibitors rescue multiple disease-causing CFTR variants
- PMID: 30753450
- PMCID: PMC6548347
- DOI: 10.1093/hmg/ddz026
HDAC inhibitors rescue multiple disease-causing CFTR variants
Abstract
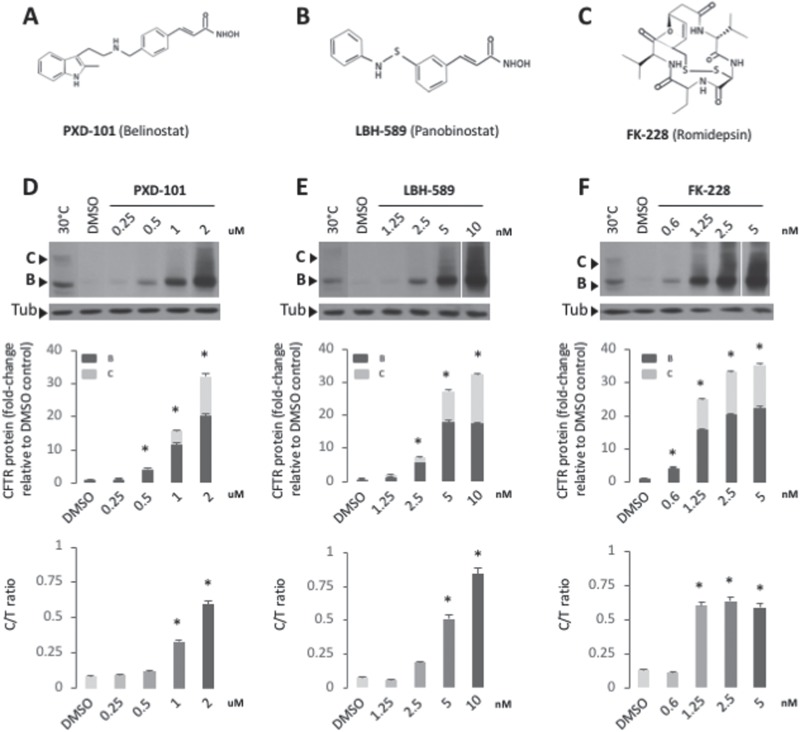
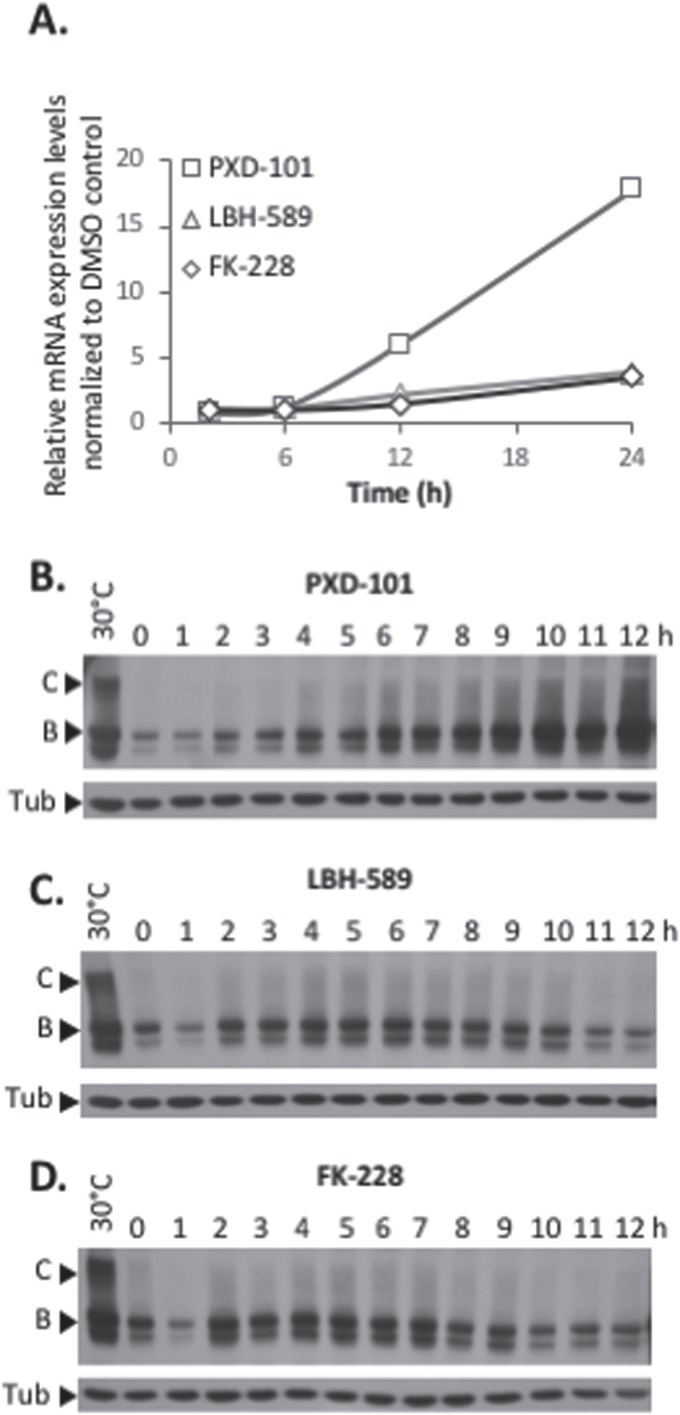
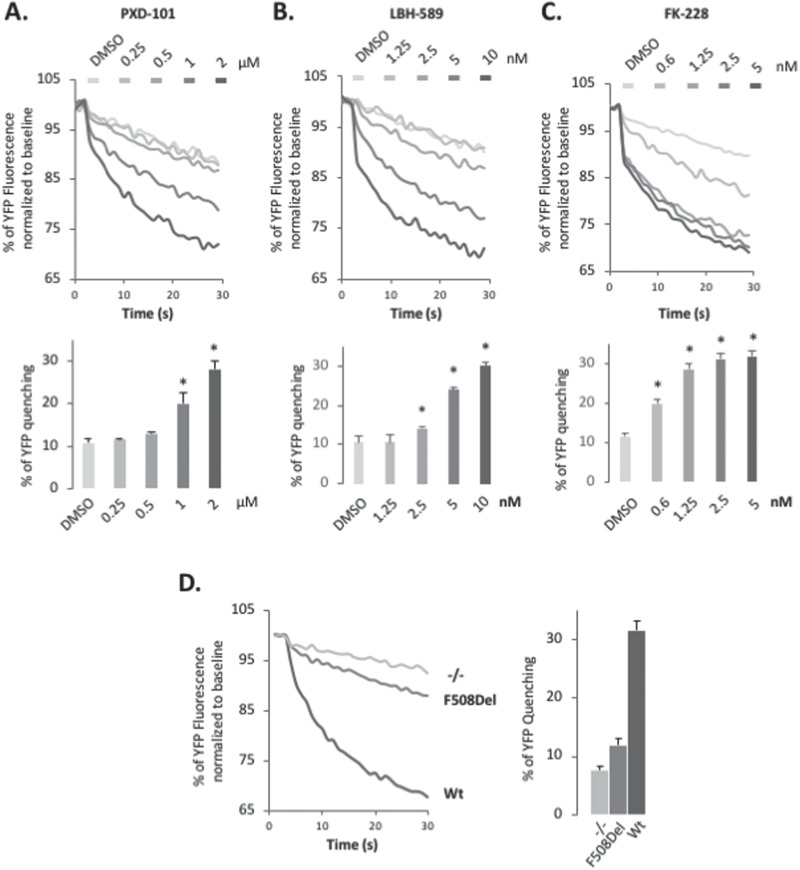
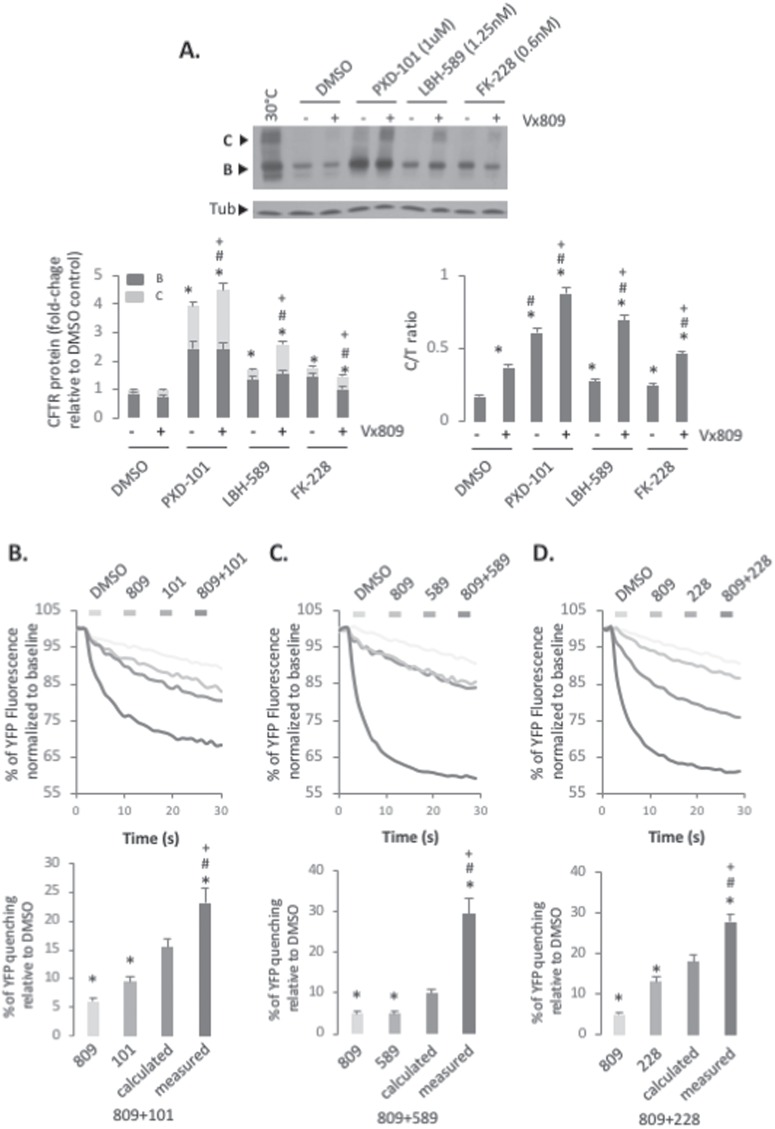
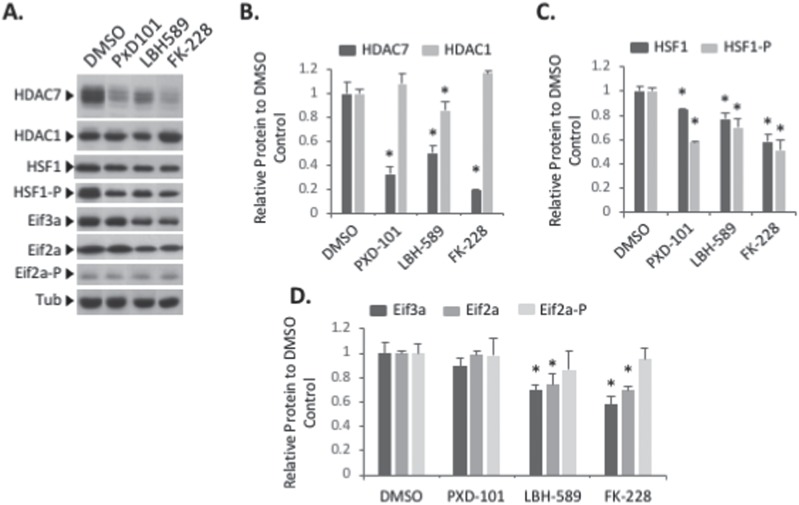
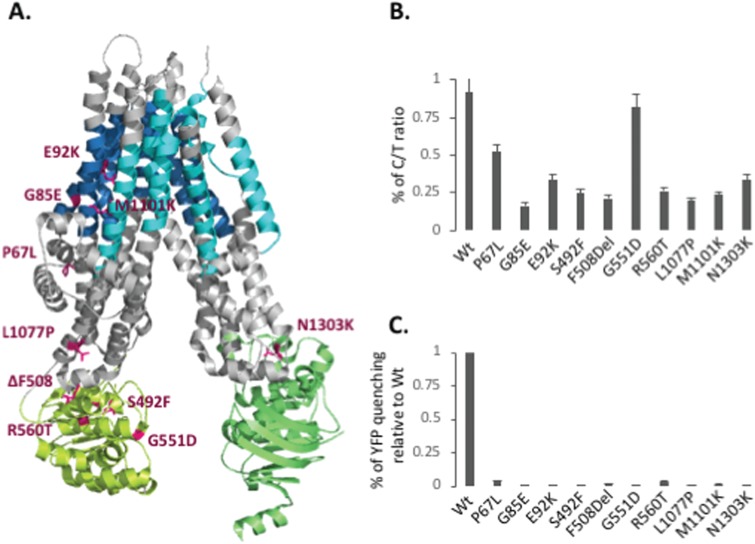
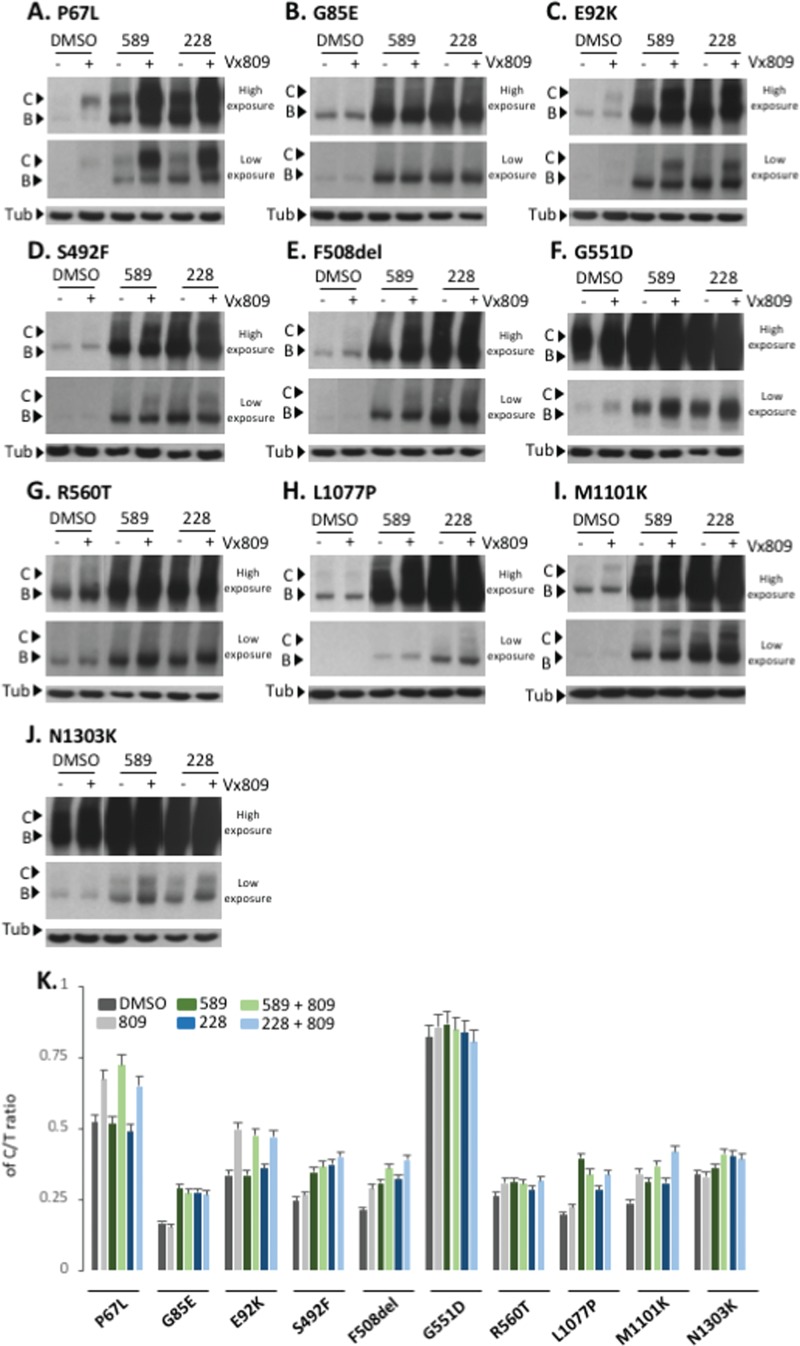
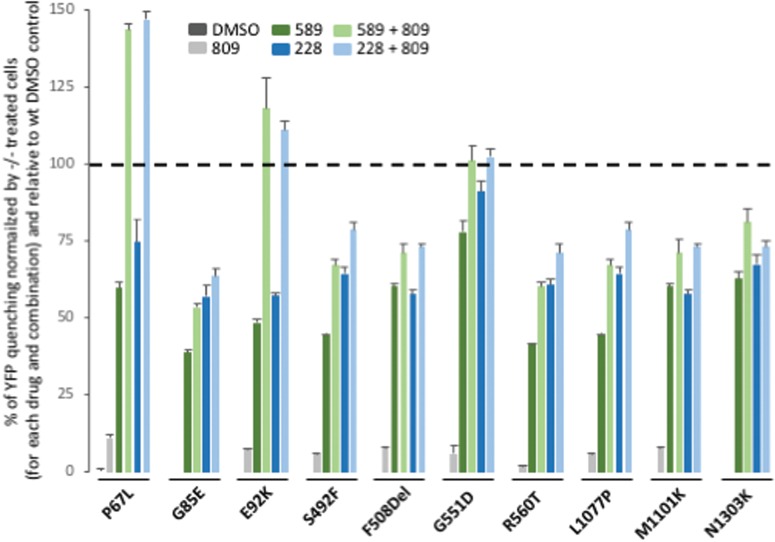
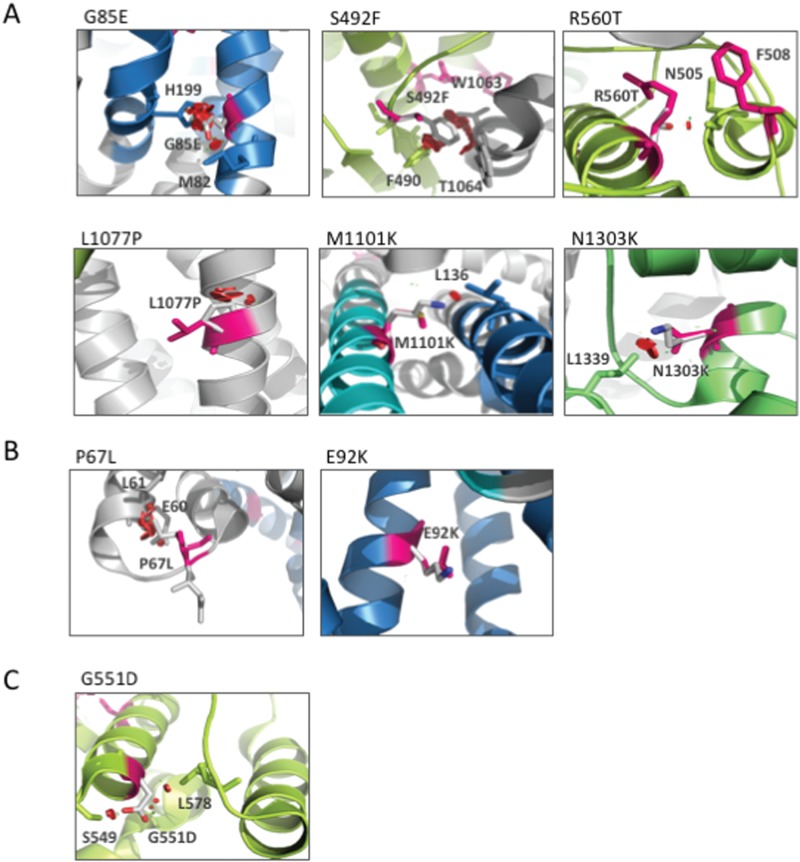
Understanding the role of the epigenome in protein-misfolding diseases remains a challenge in light of genetic diversity found in the world-wide population revealed by human genome sequencing efforts and the highly variable response of the disease population to therapeutics. An ever-growing body of evidence has shown that histone deacetylase (HDAC) inhibitors (HDACi) can have significant benefit in correcting protein-misfolding diseases that occur in response to both familial and somatic mutation. Cystic fibrosis (CF) is a familial autosomal recessive disease, caused by genetic diversity in the CF transmembrane conductance regulator (CFTR) gene, a cyclic Adenosine MonoPhosphate (cAMP)-dependent chloride channel expressed at the apical plasma membrane of epithelial cells in multiple tissues. The potential utility of HDACi in correcting the phenylalanine 508 deletion (F508del) CFTR variant as well as the over 2000 CF-associated variants remains controversial. To address this concern, we examined the impact of US Food and Drug Administration-approved HDACi on the trafficking and function of a panel of CFTR variants. Our data reveal that panobinostat (LBH-589) and romidepsin (FK-228) provide functional correction of Class II and III CFTR variants, restoring cell surface chloride channel activity in primary human bronchial epithelial cells. We further demonstrate a synergistic effect of these HDACi with Vx809, which can significantly restore channel activity for multiple CFTR variants. These data suggest that HDACi can serve to level the cellular playing field for correcting CF-causing mutations, a leveling effect that might also extend to other protein-misfolding diseases.
© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures

Similar articles
-
The HDAC inhibitor SAHA does not rescue CFTR membrane expression in Cystic Fibrosis.Int J Biochem Cell Biol. 2017 Jul;88:124-132. doi: 10.1016/j.biocel.2017.05.002. Epub 2017 May 3. Int J Biochem Cell Biol. 2017. PMID: 28478266
-
NBD2 Is Required for the Rescue of Mutant F508del CFTR by a Thiazole-Based Molecule: A Class II Corrector for the Multi-Drug Therapy of Cystic Fibrosis.Biomolecules. 2021 Sep 28;11(10):1417. doi: 10.3390/biom11101417. Biomolecules. 2021. PMID: 34680050 Free PMC article.
-
Unravelling the Regions of Mutant F508del-CFTR More Susceptible to the Action of Four Cystic Fibrosis Correctors.Int J Mol Sci. 2019 Nov 1;20(21):5463. doi: 10.3390/ijms20215463. Int J Mol Sci. 2019. PMID: 31683989 Free PMC article.
-
Lumacaftor and ivacaftor in the management of patients with cystic fibrosis: current evidence and future prospects.Ther Adv Respir Dis. 2015 Dec;9(6):313-26. doi: 10.1177/1753465815601934. Epub 2015 Sep 28. Ther Adv Respir Dis. 2015. PMID: 26416827 Review.
-
F508del-cystic fibrosis transmembrane regulator correctors for treatment of cystic fibrosis: a patent review.Expert Opin Ther Pat. 2015;25(9):991-1002. doi: 10.1517/13543776.2015.1045878. Epub 2015 May 15. Expert Opin Ther Pat. 2015. PMID: 25971311 Review.
Cited by
-
Proteostasis Regulators in Cystic Fibrosis: Current Development and Future Perspectives.J Med Chem. 2022 Apr 14;65(7):5212-5243. doi: 10.1021/acs.jmedchem.1c01897. Epub 2022 Apr 4. J Med Chem. 2022. PMID: 35377645 Free PMC article. Review.
-
Dual Blockade of Misfolded Alpha-Sarcoglycan Degradation by Bortezomib and Givinostat Combination.Front Pharmacol. 2022 Apr 27;13:856804. doi: 10.3389/fphar.2022.856804. eCollection 2022. Front Pharmacol. 2022. PMID: 35571097 Free PMC article.
-
Beyond Mendelian Inheritance: Genetic Buffering and Phenotype Variability.Phenomics. 2021 Dec 27;2(2):79-87. doi: 10.1007/s43657-021-00030-1. eCollection 2022 Apr. Phenomics. 2021. PMID: 36939776 Free PMC article. Review.
-
Spatial covariance analysis reveals the residue-by-residue thermodynamic contribution of variation to the CFTR fold.Commun Biol. 2022 Apr 13;5(1):356. doi: 10.1038/s42003-022-03302-2. Commun Biol. 2022. PMID: 35418593 Free PMC article.
-
Triangulating variation in the population to define mechanisms for precision management of genetic disease.Structure. 2022 Aug 4;30(8):1190-1207.e5. doi: 10.1016/j.str.2022.05.011. Epub 2022 Jun 16. Structure. 2022. PMID: 35714602 Free PMC article.
References
-
- Hoffmann N., Lee B., Hentzer M., Rasmussen T.B., Song Z., Johansen H.K., Givskov M. and Høiby N. (2007) Azithromycin blocks quorum sensing and alginate polymer formation and increases the sensitivity to serum and stationary-growth-phase killing of Pseudomonas aeruginosa and attenuates chronic P. aeruginosa lung infection in Cftr−/− mice. Antimicrob. Agents Chemother., 51, 3677–3687. - PMC - PubMed
-
- Qu B.-H., Strickland E.H. and Thomas P.J. (1997) Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J. Biol. Chem., 272, 15739–15744. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
