

CPPred: coding potential prediction based on the global description of RNA sequence
- PMID: 30753596
- PMCID: PMC6486542
- DOI: 10.1093/nar/gkz087
CPPred: coding potential prediction based on the global description of RNA sequence
Abstract
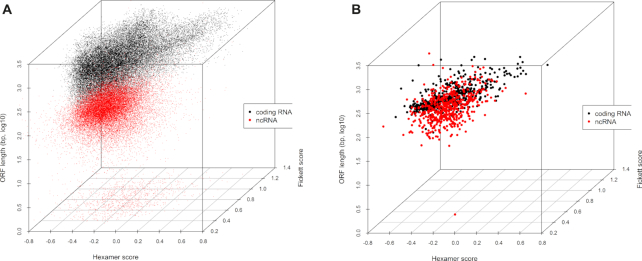
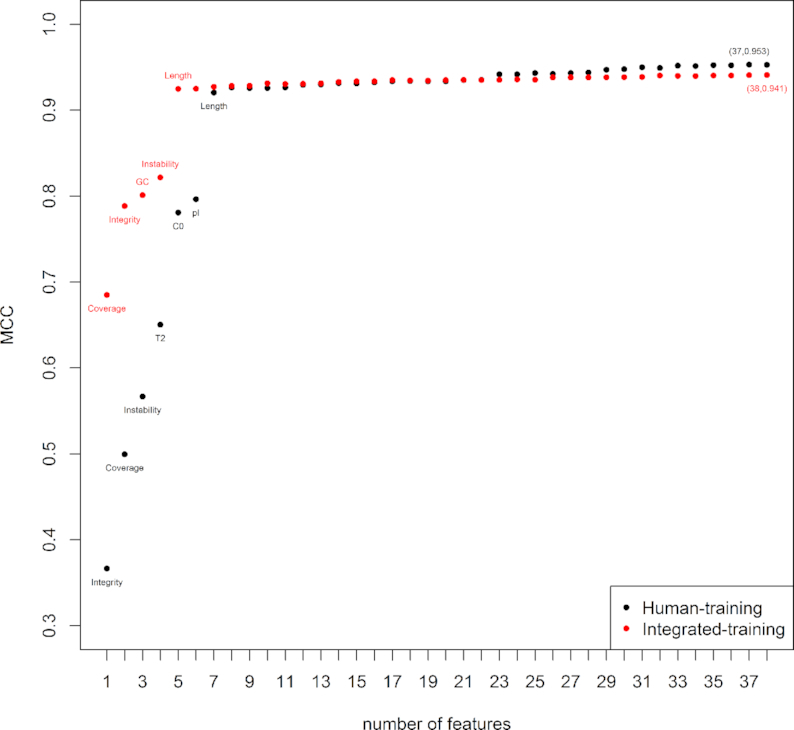
The rapid and accurate approach to distinguish between coding RNAs and ncRNAs has been playing a critical role in analyzing thousands of novel transcripts, which have been generated in recent years by next-generation sequencing technology. Previously developed methods CPAT, CPC2 and PLEK can distinguish coding RNAs and ncRNAs very well, but poorly distinguish between small coding RNAs and small ncRNAs. Herein, we report an approach, CPPred (coding potential prediction), which is based on SVM classifier and multiple sequence features including novel RNA features encoded by the global description. The CPPred can better distinguish not only between coding RNAs and ncRNAs, but also between small coding RNAs and small ncRNAs than the state-of-the-art methods due to the addition of the novel RNA features. A recent study proposes 1335 novel human coding RNAs from a large number of RNA-seq datasets. However, only 119 transcripts are predicted as coding RNAs by the CPPred. In fact, almost all proposed novel coding RNAs are ncRNAs (91.1%), which is consistent with previous reports. Remarkably, we also reveal that the global description of encoding features (T2, C0 and GC) plays an important role in the prediction of coding potential.
© The Author(s) 2019. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures


References
-
- Wang Y., Li Y., Wang Q., Lv Y., Wang S., Chen X., Yu X., Jiang W., Li X.. Computational identification of human long intergenic non-coding RNAs using a GA-SVM algorithm. Gene. 2014; 533:94–99. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
