

ACBD3 Is an Essential Pan-enterovirus Host Factor That Mediates the Interaction between Viral 3A Protein and Cellular Protein PI4KB
- PMID: 30755512
- PMCID: PMC6372799
- DOI: 10.1128/mBio.02742-18
ACBD3 Is an Essential Pan-enterovirus Host Factor That Mediates the Interaction between Viral 3A Protein and Cellular Protein PI4KB
Abstract
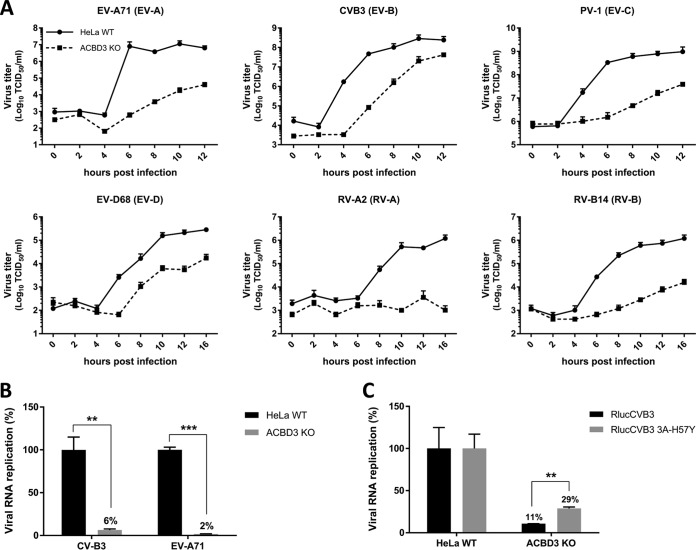
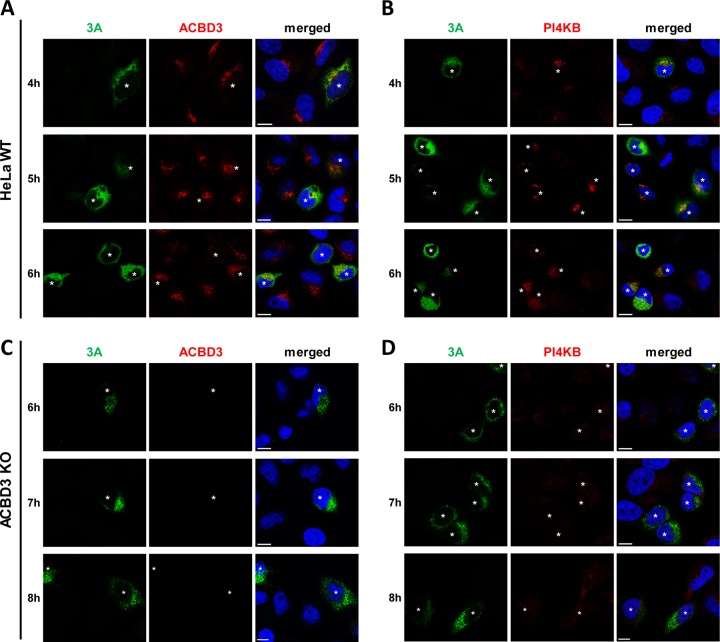
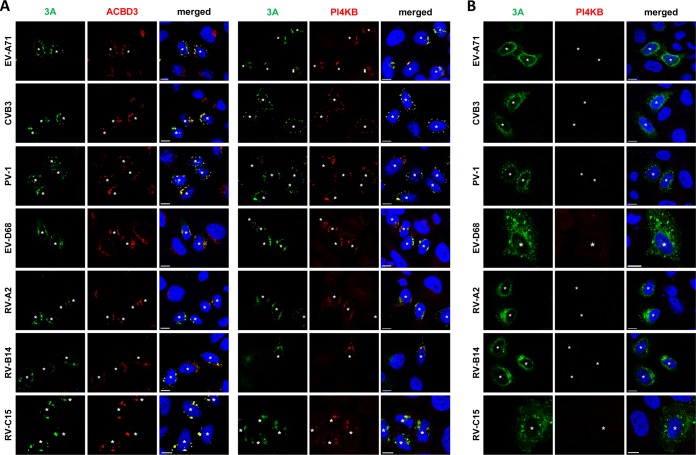
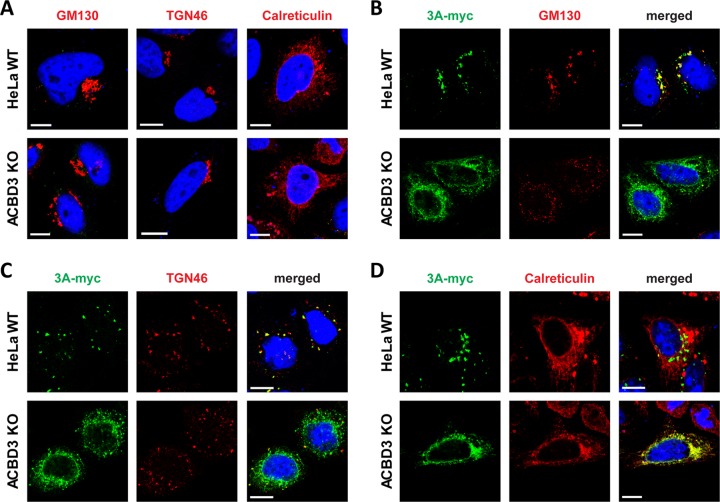
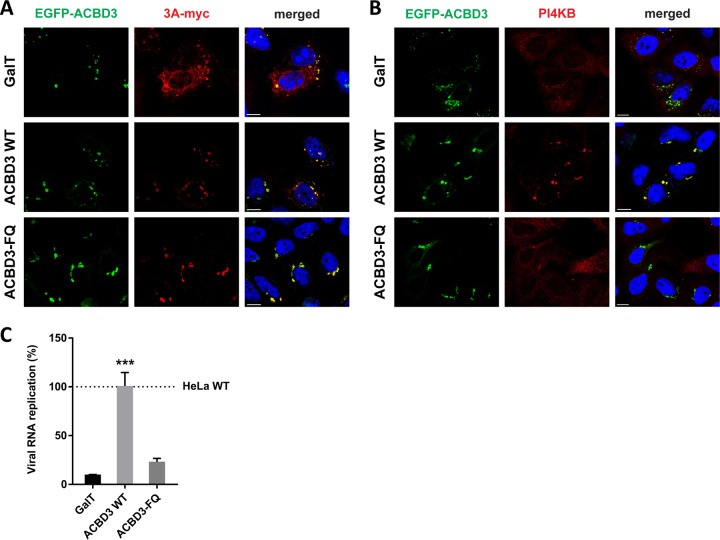
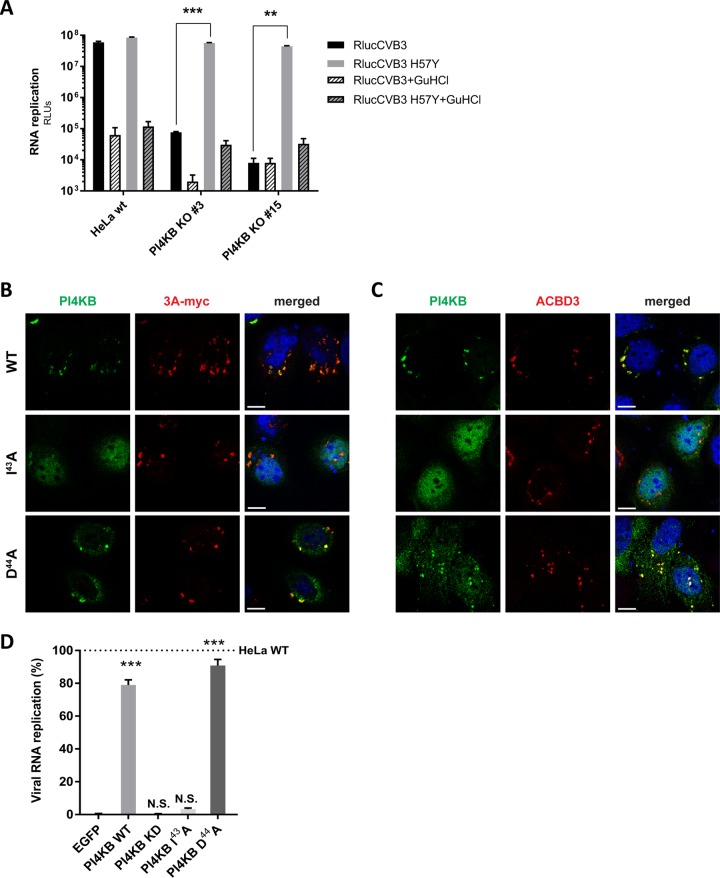
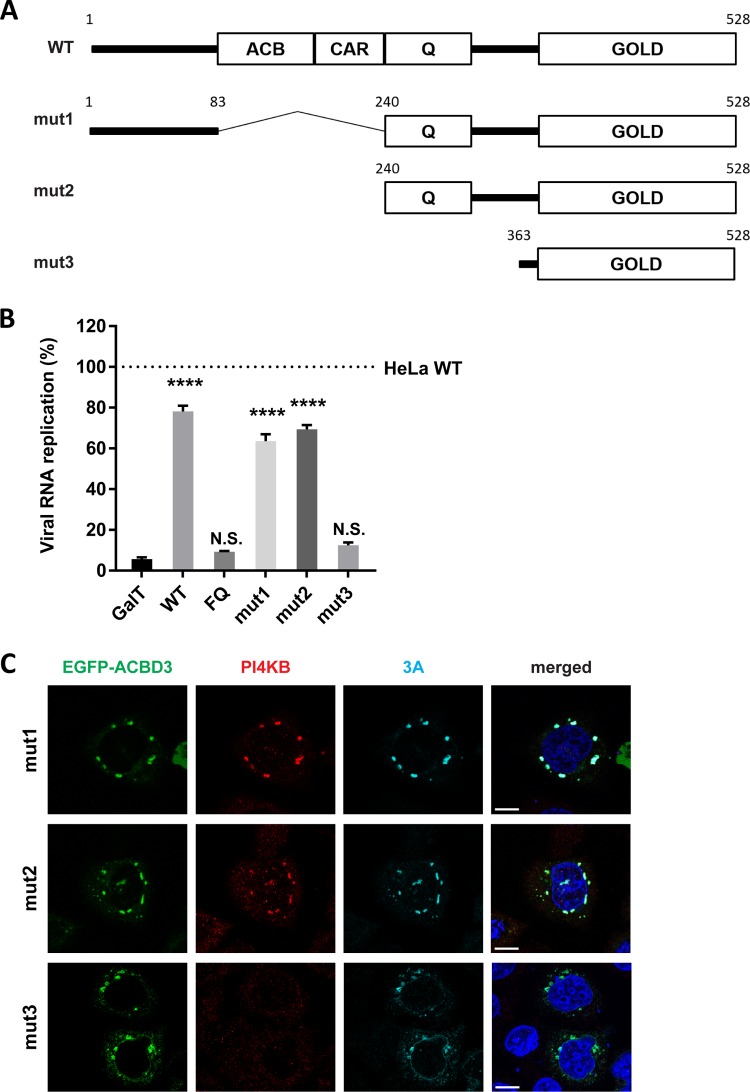
The enterovirus genus of the picornavirus family includes a large number of important human pathogens such as poliovirus, coxsackievirus, enterovirus A71, and rhinoviruses. Like all other positive-strand RNA viruses, genome replication of enteroviruses occurs on rearranged membranous structures called replication organelles (ROs). Phosphatidylinositol 4-kinase IIIβ (PI4KB) is required by all enteroviruses for RO formation. The enteroviral 3A protein recruits PI4KB to ROs, but the exact mechanism remains elusive. Here, we investigated the role of acyl-coenzyme A binding domain containing 3 (ACBD3) in PI4KB recruitment upon enterovirus replication using ACBD3 knockout (ACBD3KO) cells. ACBD3 knockout impaired replication of representative viruses from four enterovirus species and two rhinovirus species. PI4KB recruitment was not observed in the absence of ACBD3. The lack of ACBD3 also affected the localization of individually expressed 3A, causing 3A to localize to the endoplasmic reticulum instead of the Golgi. Reconstitution of wild-type (wt) ACBD3 restored PI4KB recruitment and 3A localization, while an ACBD3 mutant that cannot bind to PI4KB restored 3A localization, but not virus replication. Consistently, reconstitution of a PI4KB mutant that cannot bind ACBD3 failed to restore virus replication in PI4KBKO cells. Finally, by reconstituting ACBD3 mutants lacking specific domains in ACBD3KO cells, we show that acyl-coenzyme A binding (ACB) and charged-amino-acid region (CAR) domains are dispensable for 3A-mediated PI4KB recruitment and efficient enterovirus replication. Altogether, our data provide new insight into the central role of ACBD3 in recruiting PI4KB by enterovirus 3A and reveal the minimal domains of ACBD3 involved in recruiting PI4KB and supporting enterovirus replication.IMPORTANCE Similar to all other positive-strand RNA viruses, enteroviruses reorganize host cellular membranes for efficient genome replication. A host lipid kinase, PI4KB, plays an important role in this membrane rearrangement. The exact mechanism of how enteroviruses recruit PI4KB was unclear. Here, we revealed a role of a Golgi-residing protein, ACBD3, as a mediator of PI4KB recruitment upon enterovirus replication. ACBD3 is responsible for proper localization of enteroviral 3A proteins in host cells, which is important for 3A to recruit PI4KB. By testing ACBD3 and PI4KB mutants that abrogate the ACBD3-PI4KB interaction, we showed that this interaction is crucial for enterovirus replication. The importance of specific domains of ACBD3 was evaluated for the first time, and the domains that are essential for enterovirus replication were identified. Our findings open up a possibility for targeting ACBD3 or its interaction with enteroviruses as a novel strategy for the development of broad-spectrum antienteroviral drugs.
Keywords: acyl-coenzyme A binding domain containing 3 (ACBD3); enterovirus; phosphatidylinositol 4-kinase IIIβ (PI4KB); replication organelles; viral replication; virus-host interactions.
Copyright © 2019 Lyoo et al.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
