

Revisit Population-based and Family-based Genotype Imputation
- PMID: 30755687
- PMCID: PMC6372660
- DOI: 10.1038/s41598-018-38469-4
Revisit Population-based and Family-based Genotype Imputation
Abstract
Genome-Wide Association (GWA) with population-based imputation (PBI) has been successful in identifying common variants associated with complex diseases; however, much heritability remains to be explained and low frequency variants (LFV) may contribute. To identify LFV, a study of unrelated individuals may no longer be as efficient as a family study, where rare population variants can be frequent in families. Family-based imputation (FBI) provides an opportunity to evaluate LFV. To compare the performance of PBI and FBI, we conducted extensive simulations, generating genotypes using SeqSIMLA from various reference panels for families. We masked genotype information for variants unavailable in Framingham 550 K GWA genotype data in less informative subjects selected by GIGI-Pick. We implemented IMPUTE2 with duoHMM in SHAPEIT (Impute2_duoHMM) for PBI, MERLIN and GIGI for FBI and PedBLIMP for a hybrid approach. In general, FBI in both MERLIN and GIGI outperformed other approaches with imputation accuracy greater than 0.99 for the squared correlation and imputation quality scores (IQS) especially for LFV, although imputation accuracy from MERLIN depends on pedigree splitting for larger families. PBI performed worst with the exception of good imputation accuracy for common variants when a closely ancestry matched reference is used. In summary, linkage disequilibrium (LD) information from large available genotype resources provides good imputation for common variants with well-selected reference panels without requiring densely sequenced data in family members, while imputation of LFV with FBI benefits more from information on inheritance patterns within families yielding better imputation.
Conflict of interest statement
The authors declare no competing interests.
Figures
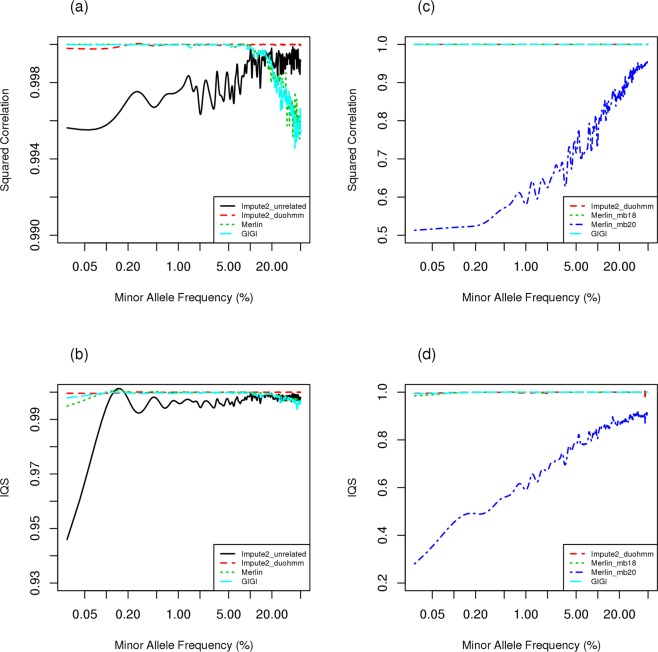
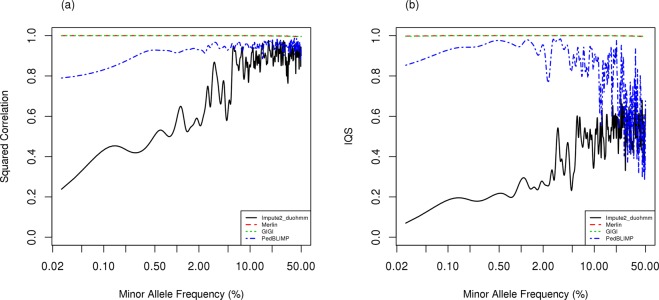
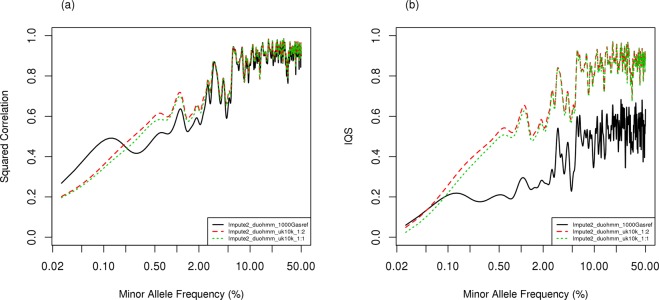
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
