

Role of mitochondrial calcium uniporter-mediated Ca2+ and iron accumulation in traumatic brain injury
- PMID: 30756474
- PMCID: PMC6433723
- DOI: 10.1111/jcmm.14206
Role of mitochondrial calcium uniporter-mediated Ca2+ and iron accumulation in traumatic brain injury
Abstract
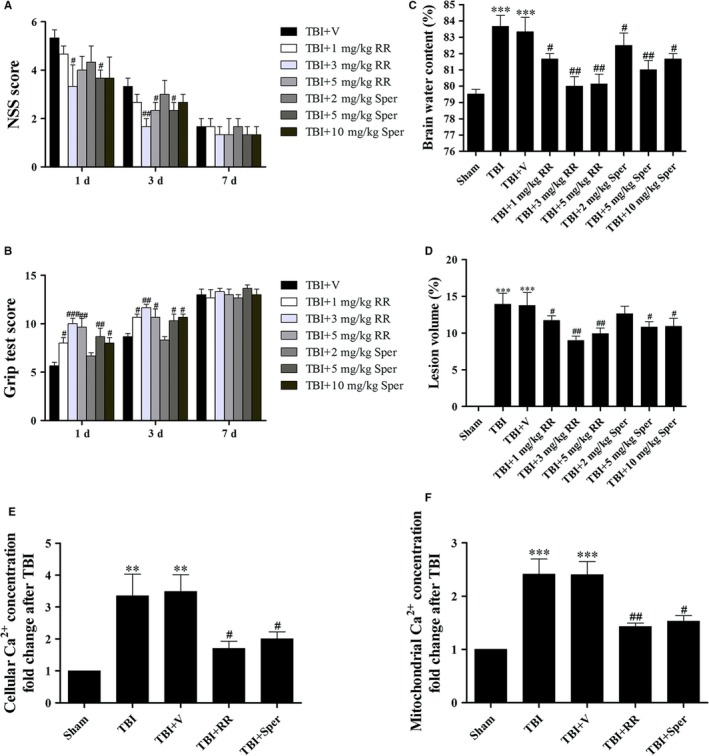
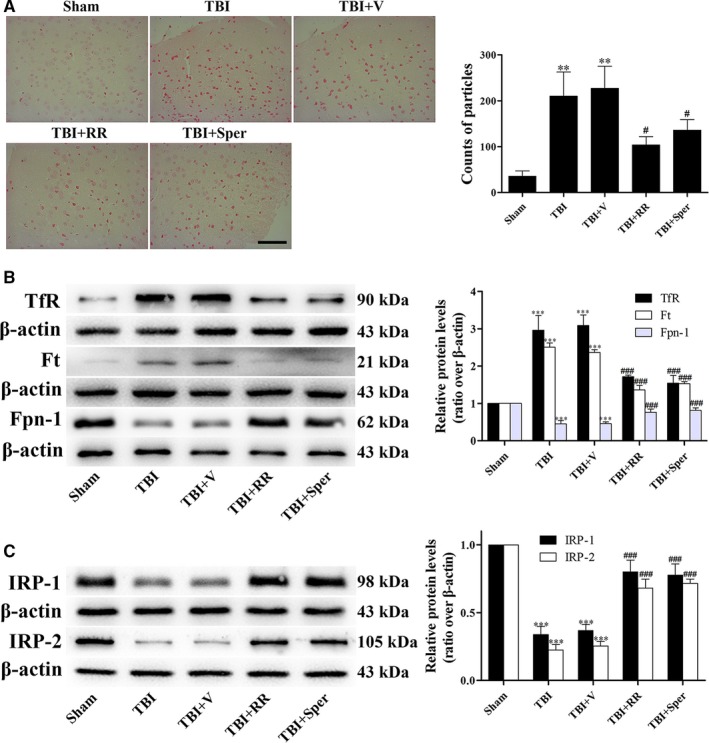
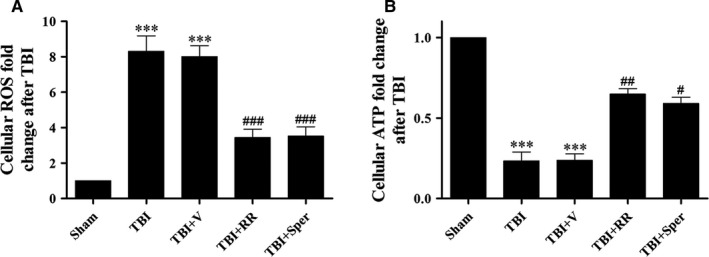
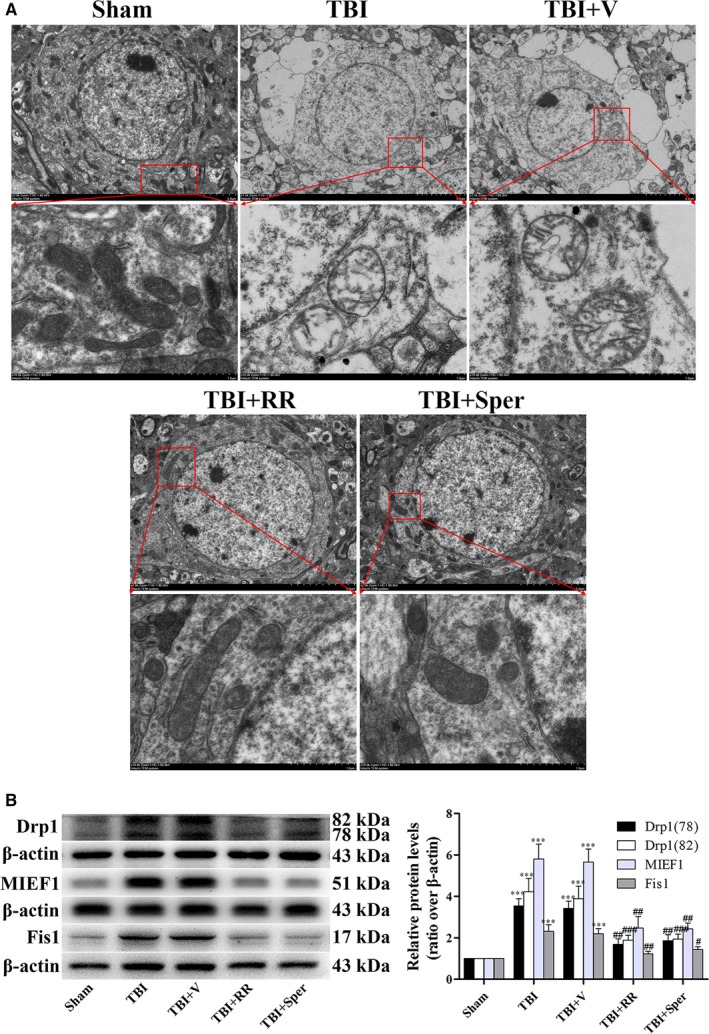
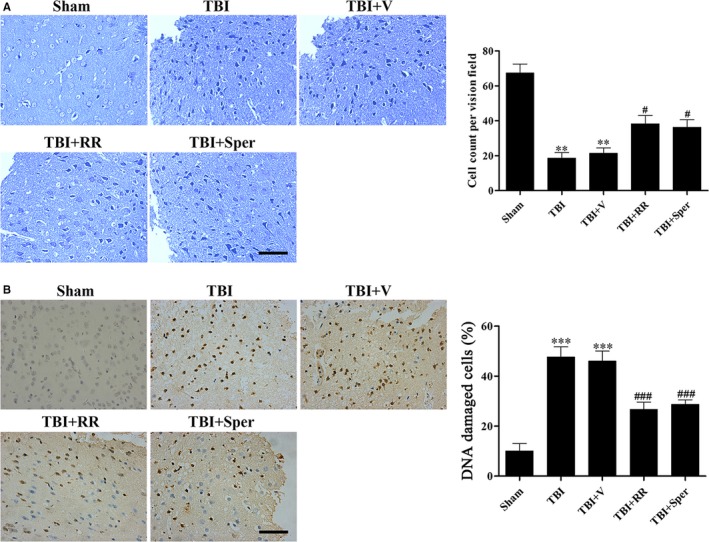
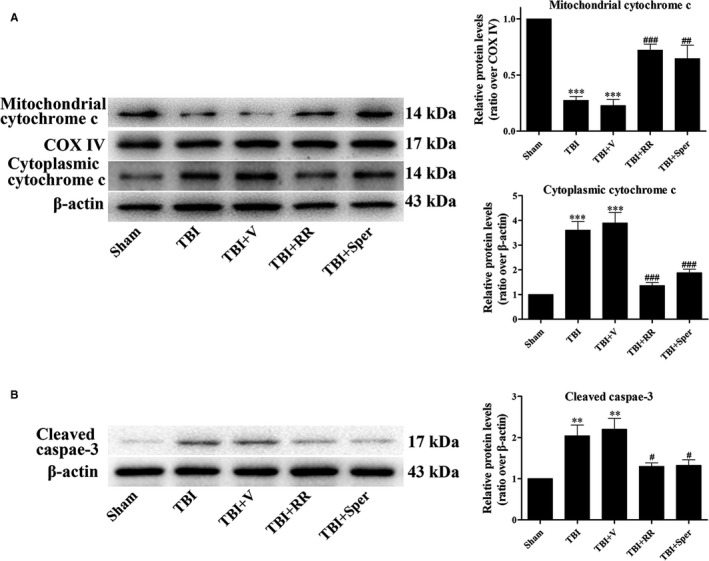
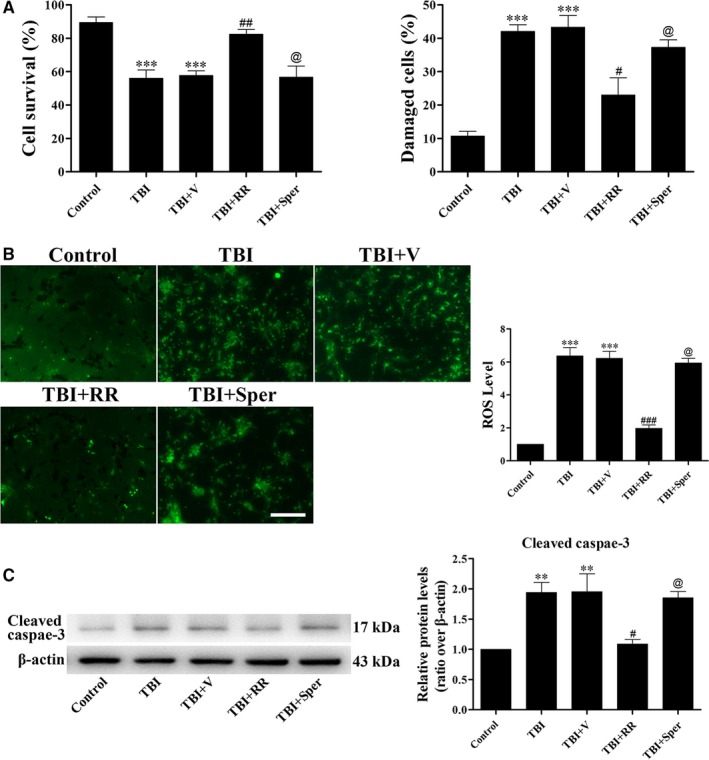
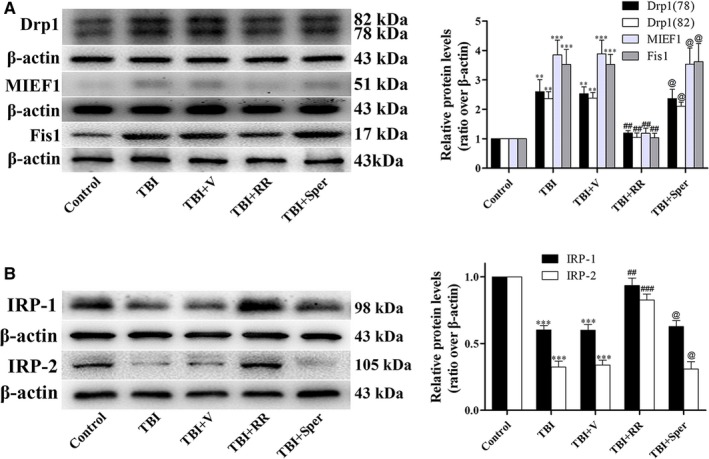
Previous studies have suggested that the cellular Ca2+ and iron homeostasis, which can be regulated by mitochondrial calcium uniporter (MCU), is associated with oxidative stress, apoptosis and many neurological diseases. However, little is known about the role of MCU-mediated Ca2+ and iron accumulation in traumatic brain injury (TBI). Under physiological conditions, MCU can be inhibited by ruthenium red (RR) and activated by spermine (Sper). In the present study, we used RR and Sper to reveal the role of MCU in mouse and neuron TBI models. Our results suggested that the Ca2+ and iron concentrations were obviously increased after TBI. In addition, TBI models showed a significant generation of reactive oxygen species (ROS), decrease in adenosine triphosphate (ATP), deformation of mitochondria, up-regulation of deoxyribonucleic acid (DNA) damage and increase in apoptosis. Blockage of MCU by RR prevented Ca2+ and iron accumulation, abated the level of oxidative stress, improved the energy supply, stabilized mitochondria, reduced DNA damage and decreased apoptosis both in vivo and in vitro. Interestingly, Sper did not increase cellular Ca2+ and iron concentrations, but suppressed the Ca2+ and iron accumulation to benefit the mice in vivo. However, Sper had no significant impact on TBI in vitro. Taken together, our data demonstrated for the first time that blockage of MCU-mediated Ca2+ and iron accumulation was essential for TBI. These findings indicated that MCU could be a novel therapeutic target for treating TBI.
Keywords: Ca2+; iron; mitochondrial calcium uniporter; neuroprotection; traumatic brain injury.
© 2019 The Authors. Journal of Cellular and Molecular Medicine published by John Wiley & Sons Ltd and Foundation for Cellular and Molecular Medicine.
Conflict of interest statement
The authors declare no potential conflicts of interest.
Figures
References
-
- Brooks JC, Strauss DJ, Shavelle RM, Paculdo DR, Hammond FM, Harrison‐Felix CL. Long‐term disability and survival in traumatic brain injury: results from the National Institute on Disability and Rehabilitation Research Model Systems. Arch Phys Med Rehabil. 2013;94:2203‐2209. - PubMed
-
- Ding K, Wang H, Xu J, et al. Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2‐ARE signaling pathway as a potential mechanism. Free Radic Biol Med. 2014;73:1‐11. - PubMed
-
- Zhang L, Wang H. Targeting the NF‐E2‐related factor 2 pathway: a novel strategy for traumatic brain injury. Mol Neurobiol. 2017. - PubMed
-
- Sun M, Zhao Y, Gu Y, Zhang Y. Protective effects of taurine against closed head injury in rats. J Neurotrauma. 2015;32:66‐74. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
