

Lysosomal Leukodystrophies Lysosomal Storage Diseases Associated With White Matter Abnormalities
- PMID: 30757954
- PMCID: PMC6459700
- DOI: 10.1177/0883073819828587
Lysosomal Leukodystrophies Lysosomal Storage Diseases Associated With White Matter Abnormalities
Abstract
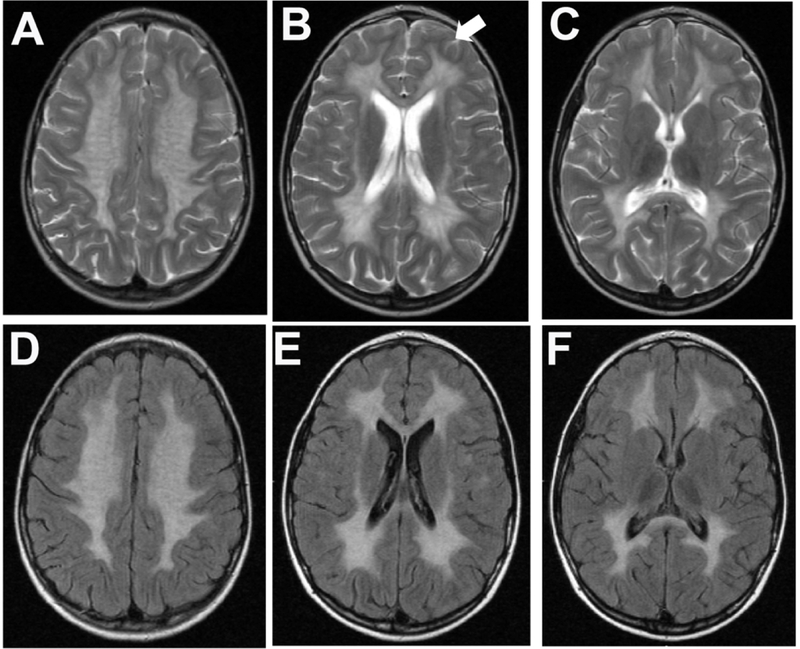
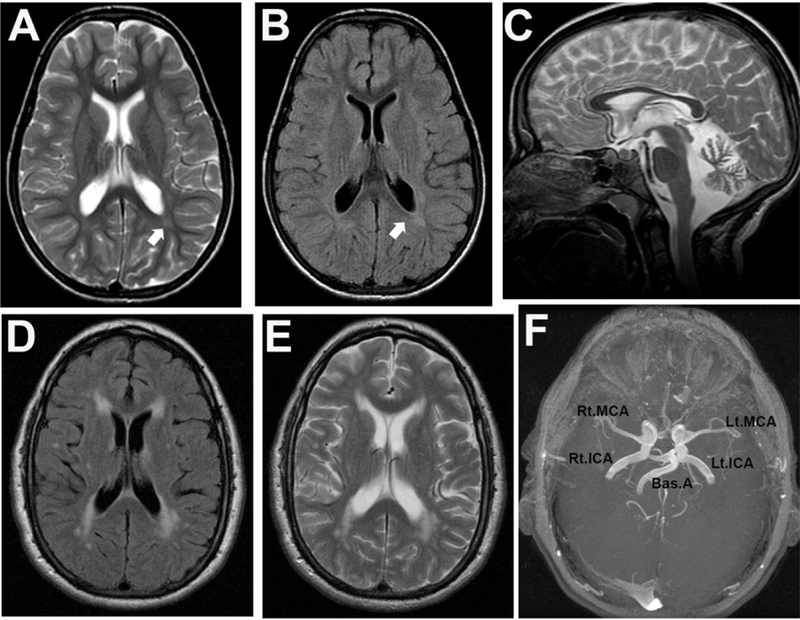
The leukodystrophies are a group of genetic metabolic diseases characterized by an abnormal development or progressive degeneration of the myelin sheath. The myelin is a complex sheath composed of several macromolecules covering axons as an insulator. Each of the leukodystrophies is caused by mutations in genes encoding enzymes that are involved in myelin production and maintenance. The lysosomal storage diseases are inborn disorders of compartmentalized cellular organelles with broad clinical manifestations secondary to the progressive accumulation of undegraded macromolecules within lysosomes and related organelles. The more than 60 different lysosomal storage diseases are rare diseases; however, collectively, the incidence of lysosomal storage diseases ranges just over 1 in 2500 live births. The majority of lysosomal storage diseases are associated with neurologic manifestations including developmental delay, seizures, acroparesthesia, motor weakness, and extrapyramidal signs. These inborn organelle disorders show wide clinical variability affecting individuals from all age groups. In addition, several of neurologic, also known as neuronopathic, lysosomal storage diseases are associated with some level of white matter disease, which often triggers the diagnostic investigation. Most lysosomal storage diseases are autosomal recessively inherited and few are X-linked, with females being at risk of presenting with mild, but clinically relevant neurologic manifestations. Biochemical assays are the basis of the diagnosis and are usually confirmed by molecular genetic testing. Novel therapies have emerged. However, most affected patients with lysosomal storage diseases have only supportive management to rely on. A better understanding of the mechanisms resulting in the leukodystrophy will certainly result in innovative and efficacious disease-modifying therapies.
Keywords: leukodystrophies; lysosomal diseases; lysosome; myelin.
Figures
Similar articles
-
Lysosomal storage diseases.Handb Clin Neurol. 2024;204:147-172. doi: 10.1016/B978-0-323-99209-1.00008-9. Handb Clin Neurol. 2024. PMID: 39322377
-
Mitochondrial Dysfunction and Neurodegeneration in Lysosomal Storage Disorders.Trends Mol Med. 2017 Feb;23(2):116-134. doi: 10.1016/j.molmed.2016.12.003. Epub 2017 Jan 19. Trends Mol Med. 2017. PMID: 28111024 Review.
-
Different molecular mechanisms leading to white matter hypomyelination in infantile onset lysosomal disorders.Neuropediatrics. 2005 Aug;36(4):265-9. doi: 10.1055/s-2005-865863. Neuropediatrics. 2005. PMID: 16138252
-
Estimating the relative frequency of leukodystrophies and recommendations for carrier screening in the era of next-generation sequencing.Am J Med Genet A. 2020 Aug;182(8):1906-1912. doi: 10.1002/ajmg.a.61641. Epub 2020 Jun 23. Am J Med Genet A. 2020. PMID: 32573057 Free PMC article.
-
CNS-Targeting Therapies for Lysosomal Storage Diseases: Current Advances and Challenges.Front Mol Biosci. 2020 Nov 12;7:559804. doi: 10.3389/fmolb.2020.559804. eCollection 2020. Front Mol Biosci. 2020. PMID: 33304924 Free PMC article. Review.
Cited by
-
Clinical Trials for Gene Therapy in Lysosomal Diseases With CNS Involvement.Front Mol Biosci. 2021 Sep 16;8:624988. doi: 10.3389/fmolb.2021.624988. eCollection 2021. Front Mol Biosci. 2021. PMID: 34604300 Free PMC article. Review.
-
Advancing CNS Therapeutics: Enhancing Neurological Disorders with Nanoparticle-Based Gene and Enzyme Replacement Therapies.Int J Nanomedicine. 2025 Feb 4;20:1443-1490. doi: 10.2147/IJN.S457393. eCollection 2025. Int J Nanomedicine. 2025. PMID: 39925682 Free PMC article. Review.
-
Artificially Induced Pluripotent Stem Cell-Derived Whole-Brain Organoid for Modelling the Pathophysiology of Metachromatic Leukodystrophy and Drug Repurposing.Biomedicines. 2021 Apr 20;9(4):440. doi: 10.3390/biomedicines9040440. Biomedicines. 2021. PMID: 33923989 Free PMC article.
-
Update on leukodystrophies and developing trials.J Neurol. 2024 Jan;271(1):593-605. doi: 10.1007/s00415-023-11996-5. Epub 2023 Sep 27. J Neurol. 2024. PMID: 37755460 Free PMC article. Review.
-
The Role of Microglia in Inherited White-Matter Disorders and Connections to Frontotemporal Dementia.Appl Clin Genet. 2021 Mar 31;14:195-207. doi: 10.2147/TACG.S245029. eCollection 2021. Appl Clin Genet. 2021. PMID: 33833548 Free PMC article. Review.
References
-
- Meikle PJ, Hopwood JJ, Clague AE and Carey WF. Prevalence of lysosomal storage disorders. JAMA 1999;281(3): 249–54. - PubMed
-
- Metz TF, Mechtler TP, Merk M, et al. Evaluation of a novel, commercially available mass spectrometry kit for newborn screening including succinylacetone without hydrazine. Clin Chim Acta 2012;413(15–16): 1259–64. - PubMed
-
- de Duve C From cytases to lysosomes. Fed Proc 1964;23: 1045–9. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
