

Emerging Diversity in Lipid-Protein Interactions
- PMID: 30758191
- PMCID: PMC6509647
- DOI: 10.1021/acs.chemrev.8b00451
Emerging Diversity in Lipid-Protein Interactions
Abstract
Membrane lipids interact with proteins in a variety of ways, ranging from providing a stable membrane environment for proteins to being embedded in to detailed roles in complicated and well-regulated protein functions. Experimental and computational advances are converging in a rapidly expanding research area of lipid-protein interactions. Experimentally, the database of high-resolution membrane protein structures is growing, as are capabilities to identify the complex lipid composition of different membranes, to probe the challenging time and length scales of lipid-protein interactions, and to link lipid-protein interactions to protein function in a variety of proteins. Computationally, more accurate membrane models and more powerful computers now enable a detailed look at lipid-protein interactions and increasing overlap with experimental observations for validation and joint interpretation of simulation and experiment. Here we review papers that use computational approaches to study detailed lipid-protein interactions, together with brief experimental and physiological contexts, aiming at comprehensive coverage of simulation papers in the last five years. Overall, a complex picture of lipid-protein interactions emerges, through a range of mechanisms including modulation of the physical properties of the lipid environment, detailed chemical interactions between lipids and proteins, and key functional roles of very specific lipids binding to well-defined binding sites on proteins. Computationally, despite important limitations, molecular dynamics simulations with current computer power and theoretical models are now in an excellent position to answer detailed questions about lipid-protein interactions.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
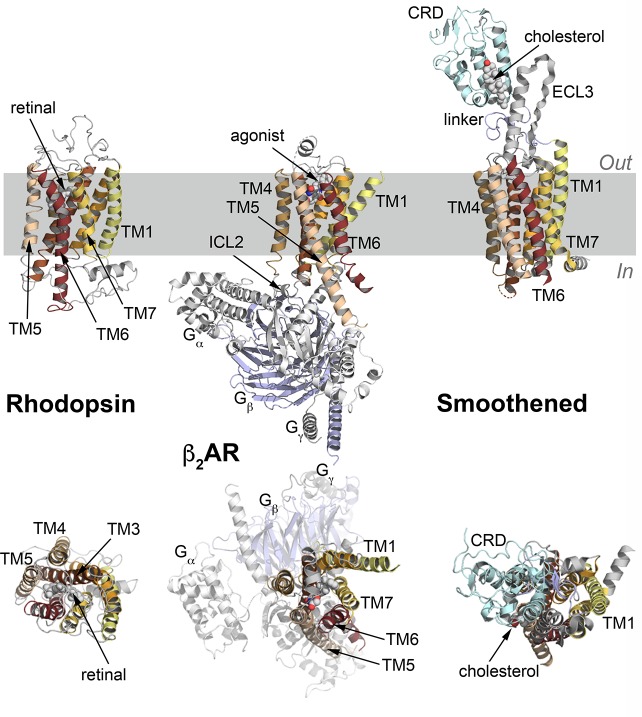
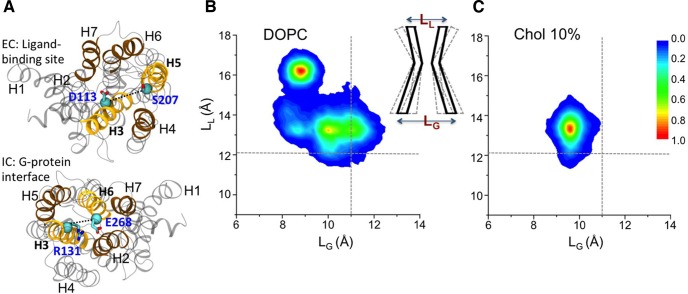
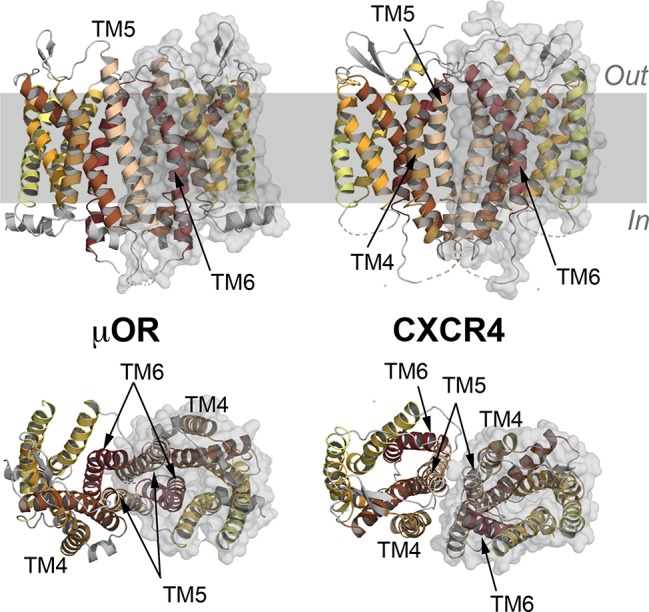
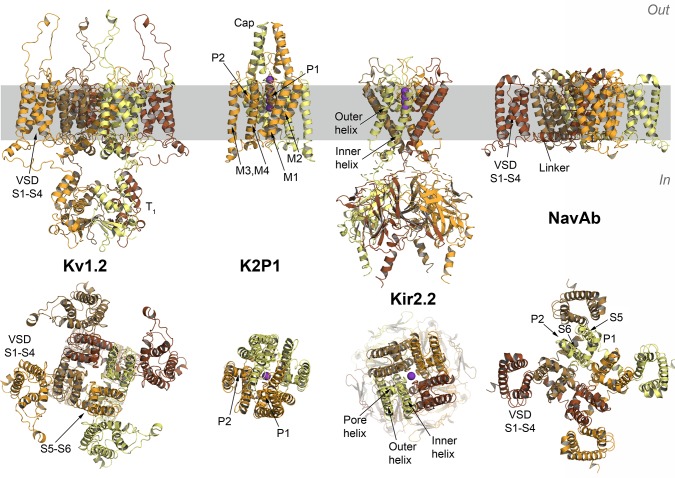
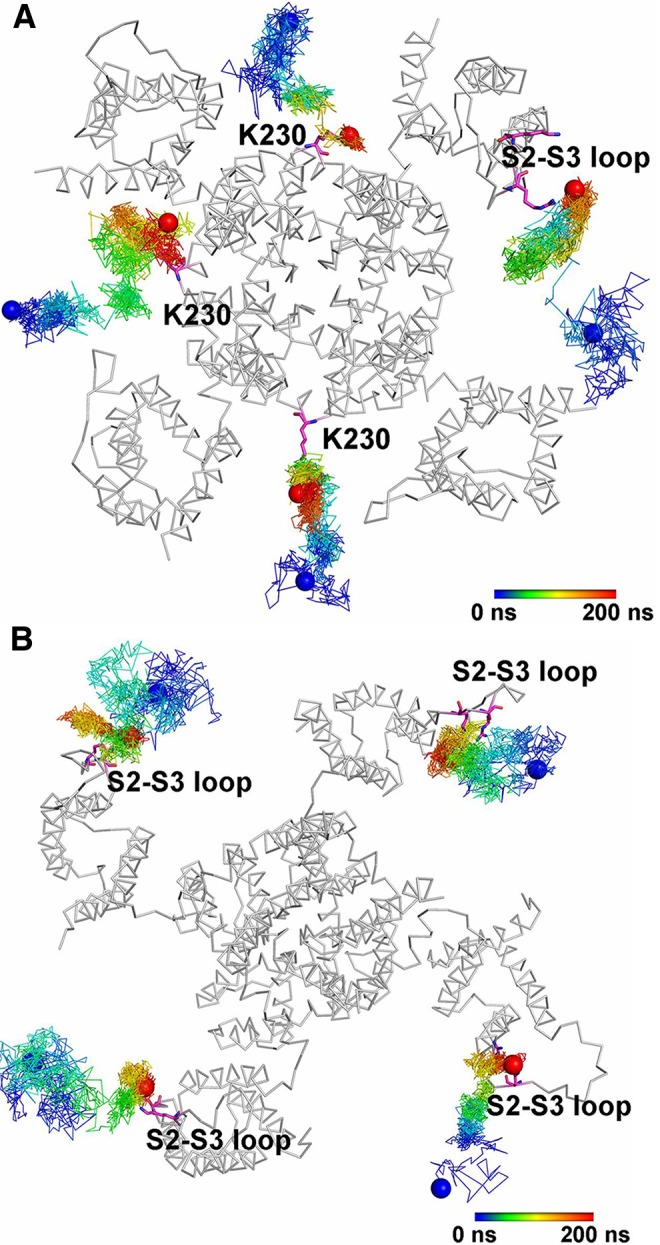
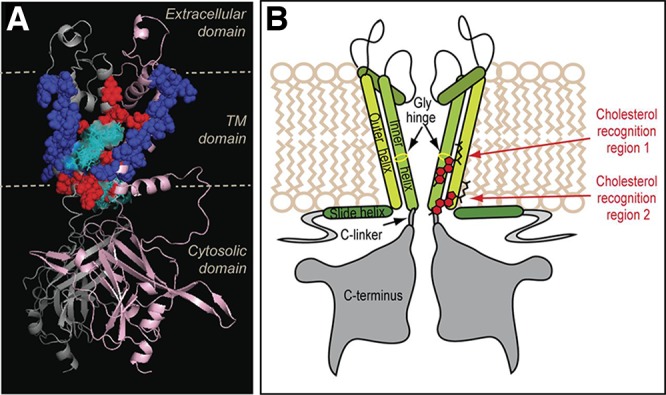
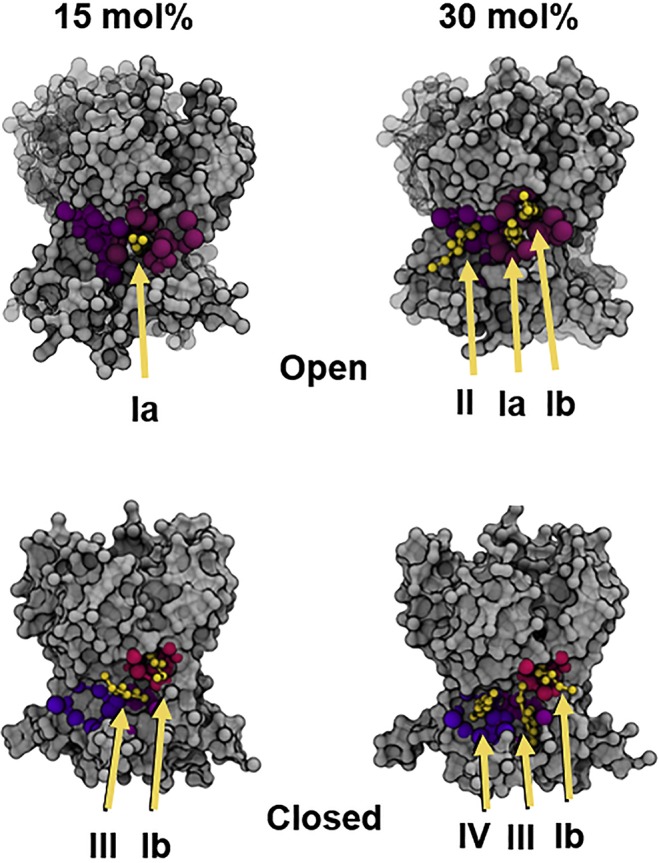
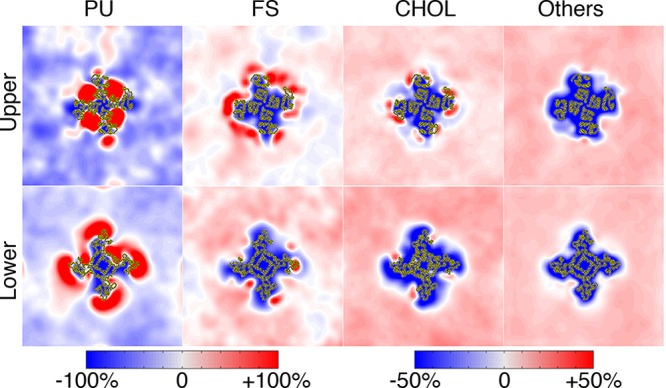
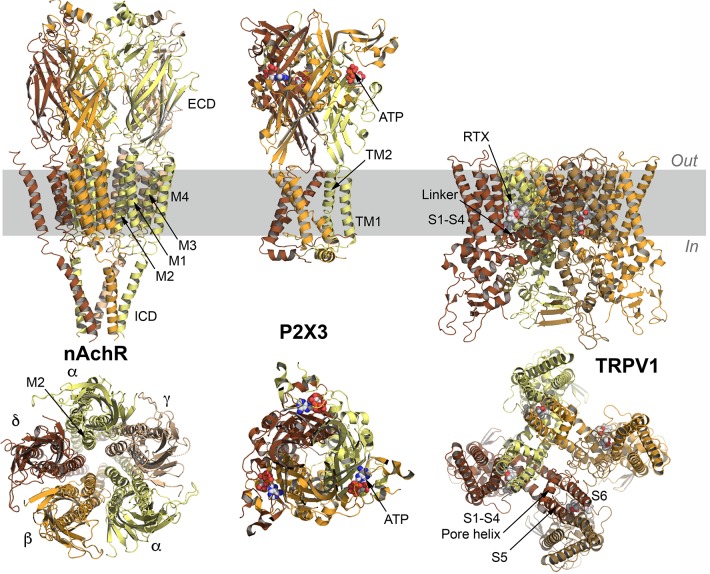
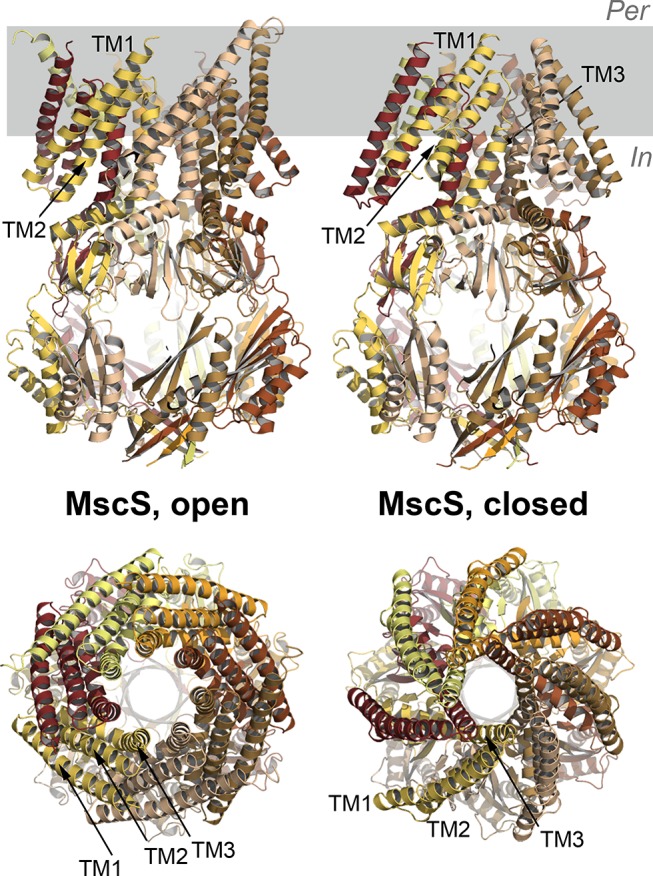
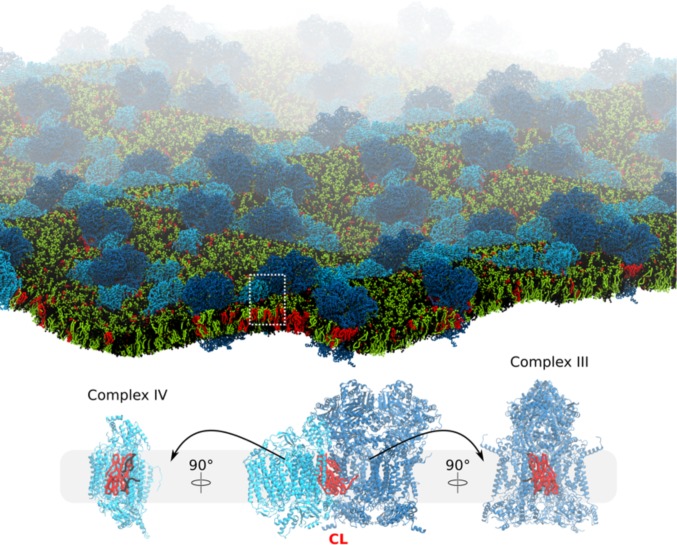
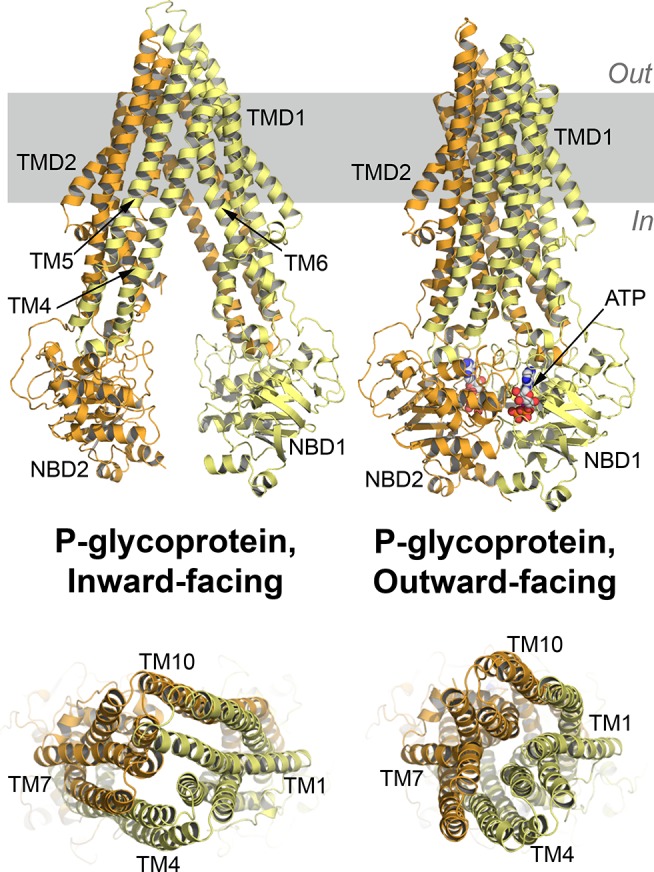

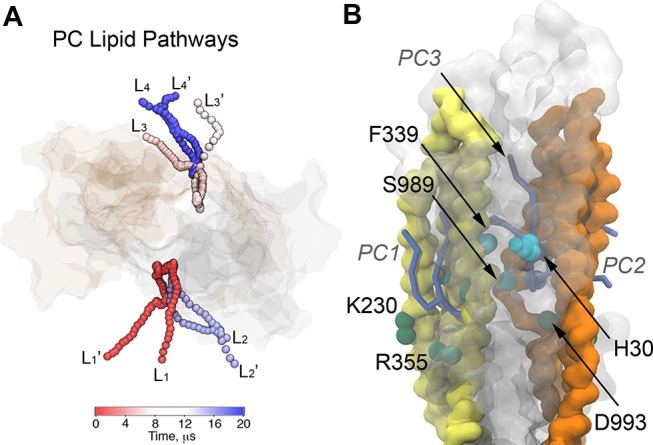
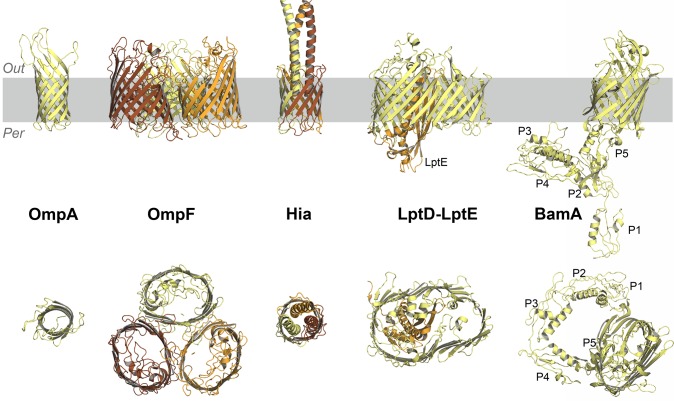
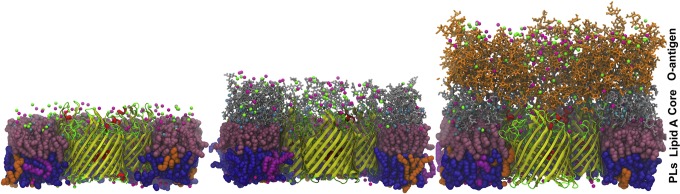
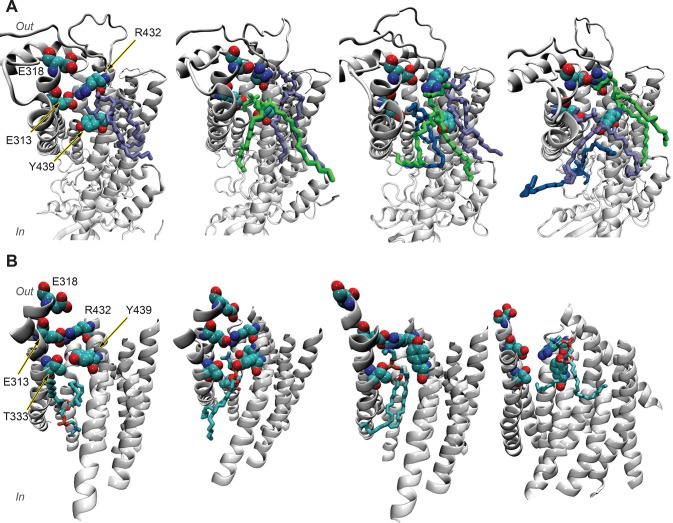
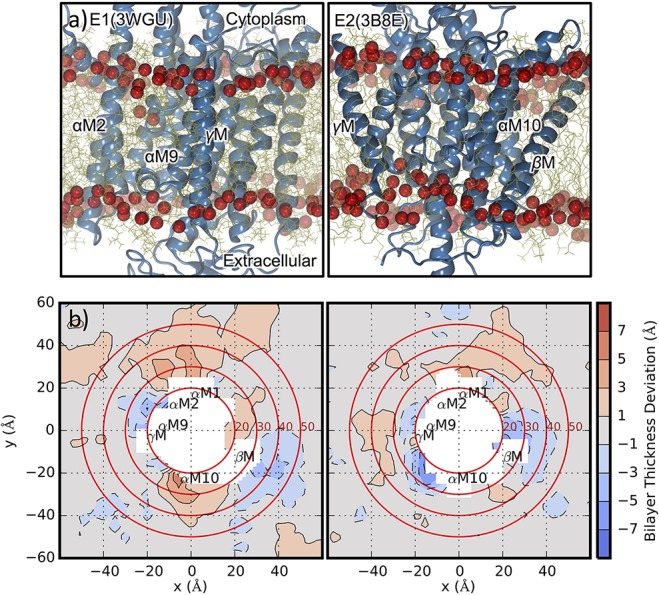
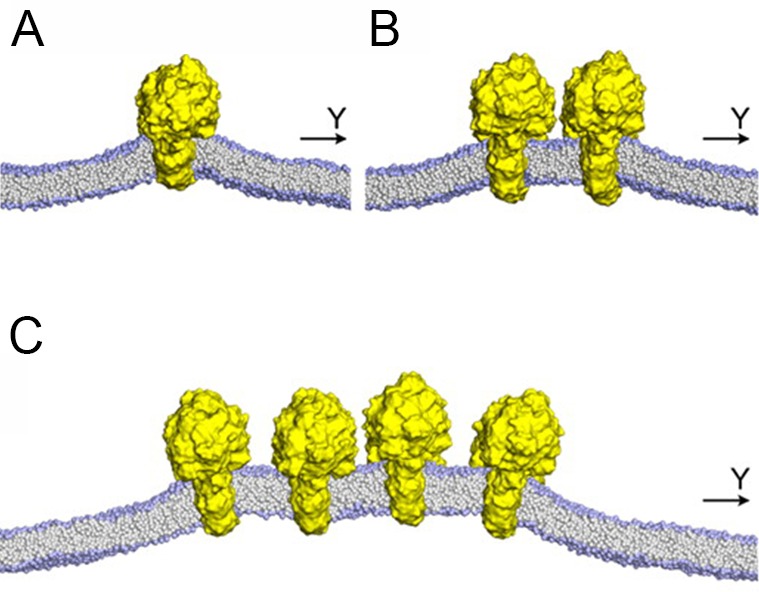
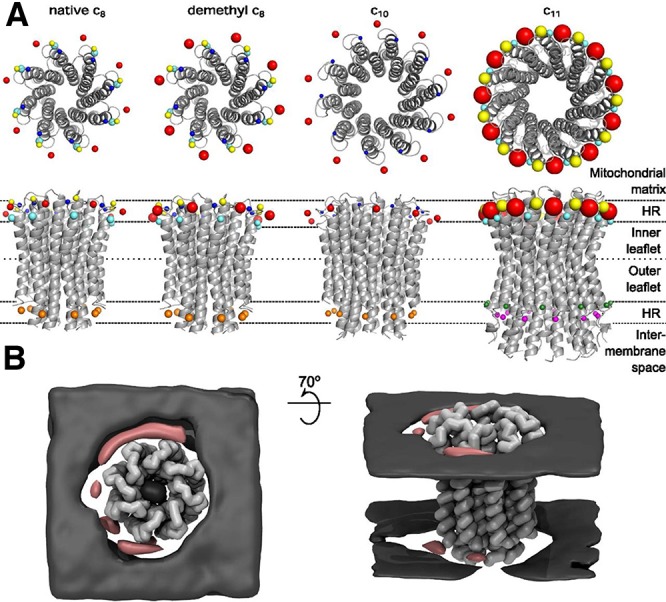
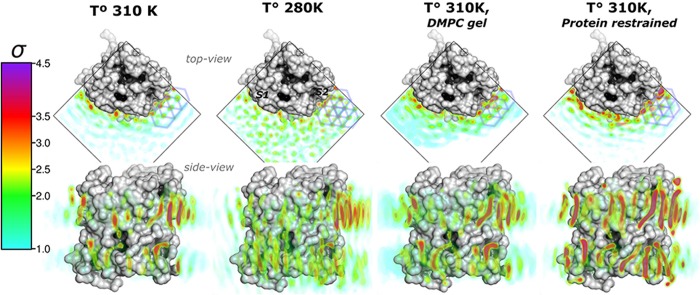
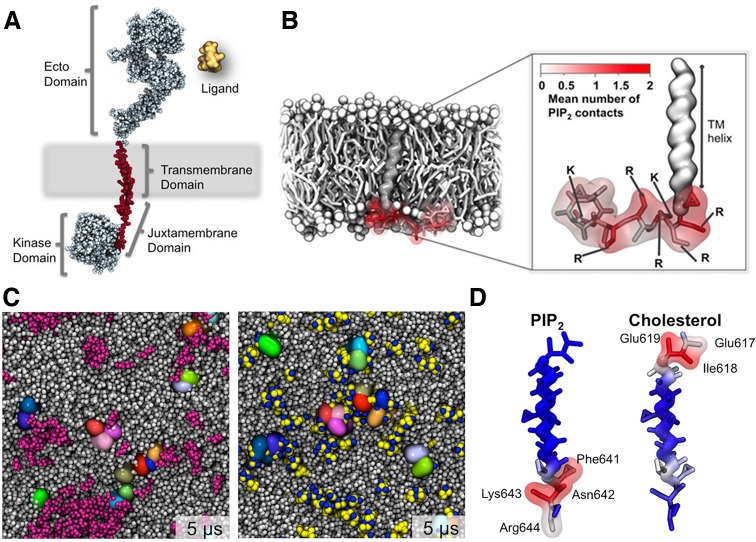
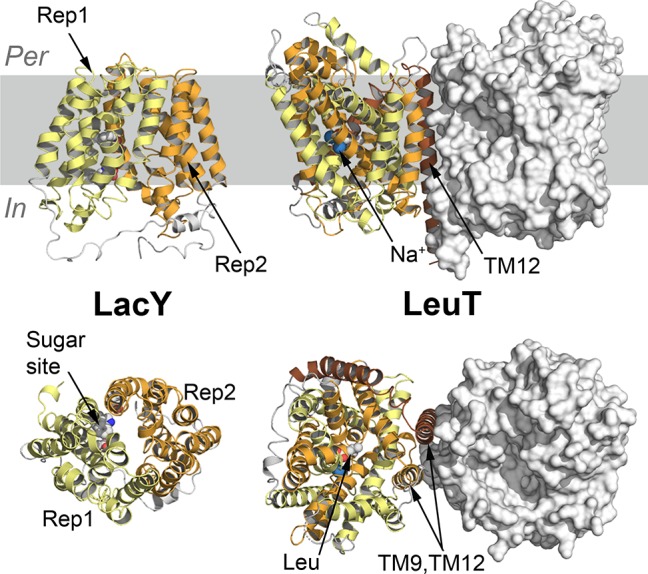
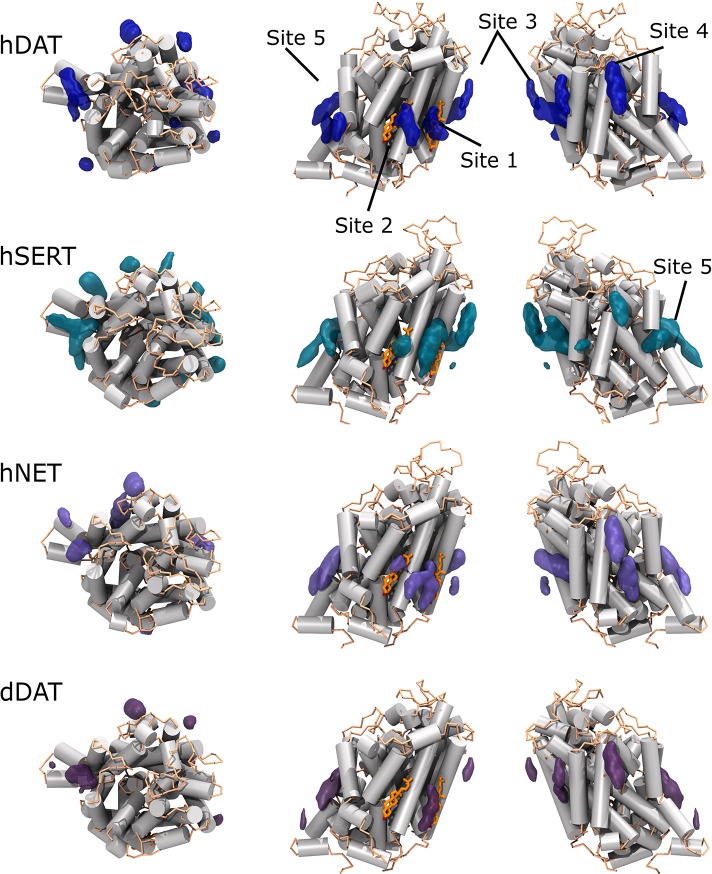
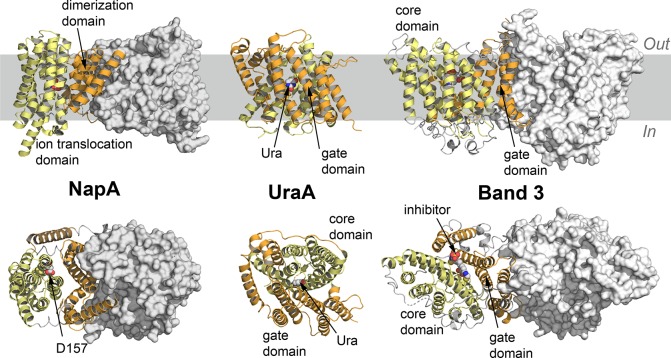
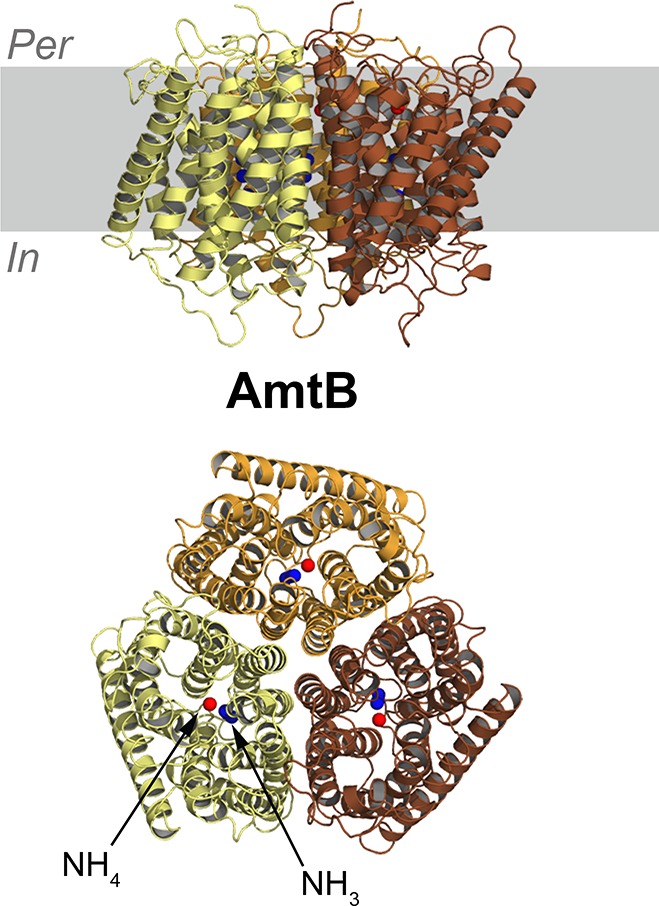
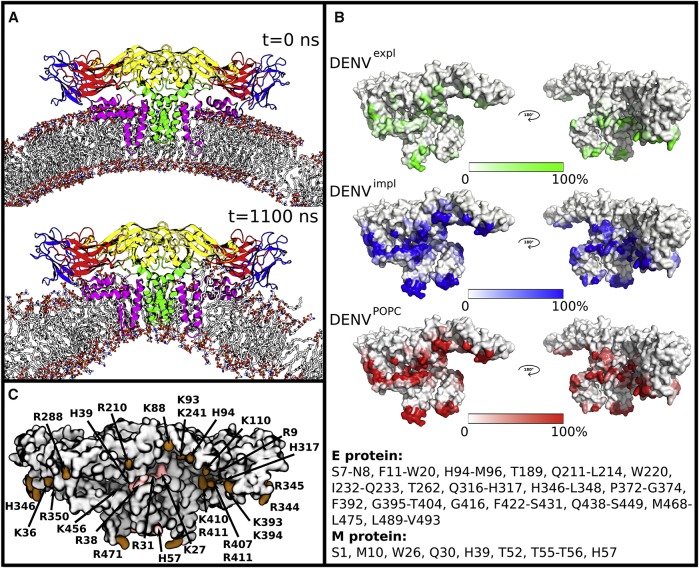
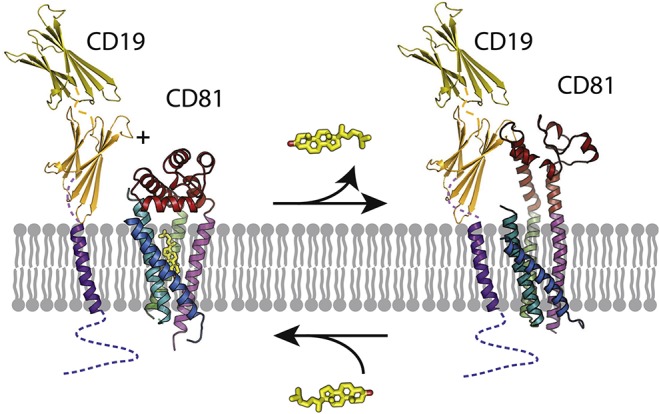
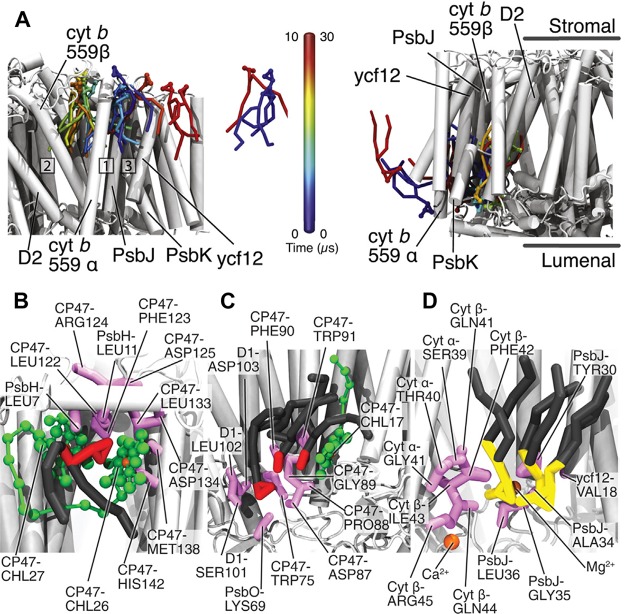
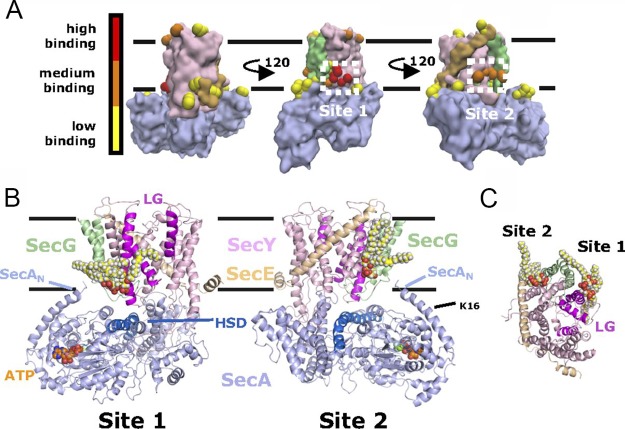
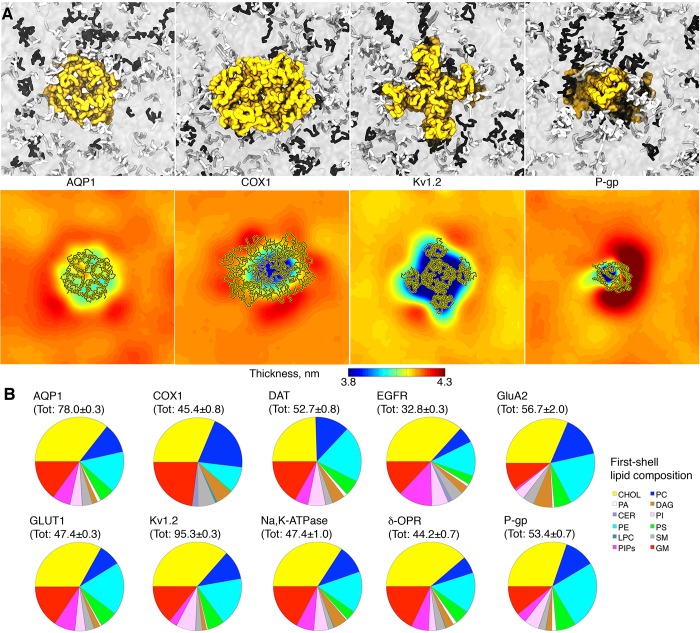
References
-
- Escriba P. V.; Gonzalez-Ros J. M.; Goni F. M.; Kinnunen P. K. J.; Vigh L.; Sanchez-Magraner L.; Fernandez A. M.; Busquets X.; Horvath I.; Barcelo-Coblijn G. Membranes: A Meeting Point for Lipids, Proteins and Therapies. J. Cell. Mol. Med. 2008, 12, 829–875. 10.1111/j.1582-4934.2008.00281.x. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
