

ERF-related craniosynostosis: The phenotypic and developmental profile of a new craniosynostosis syndrome
- PMID: 30758909
- PMCID: PMC6491982
- DOI: 10.1002/ajmg.a.61073
ERF-related craniosynostosis: The phenotypic and developmental profile of a new craniosynostosis syndrome
Abstract
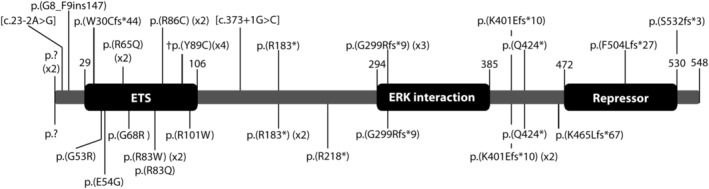
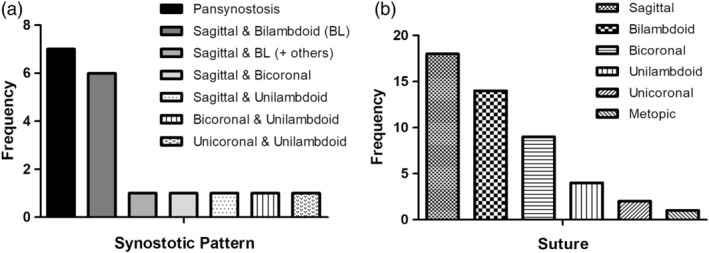
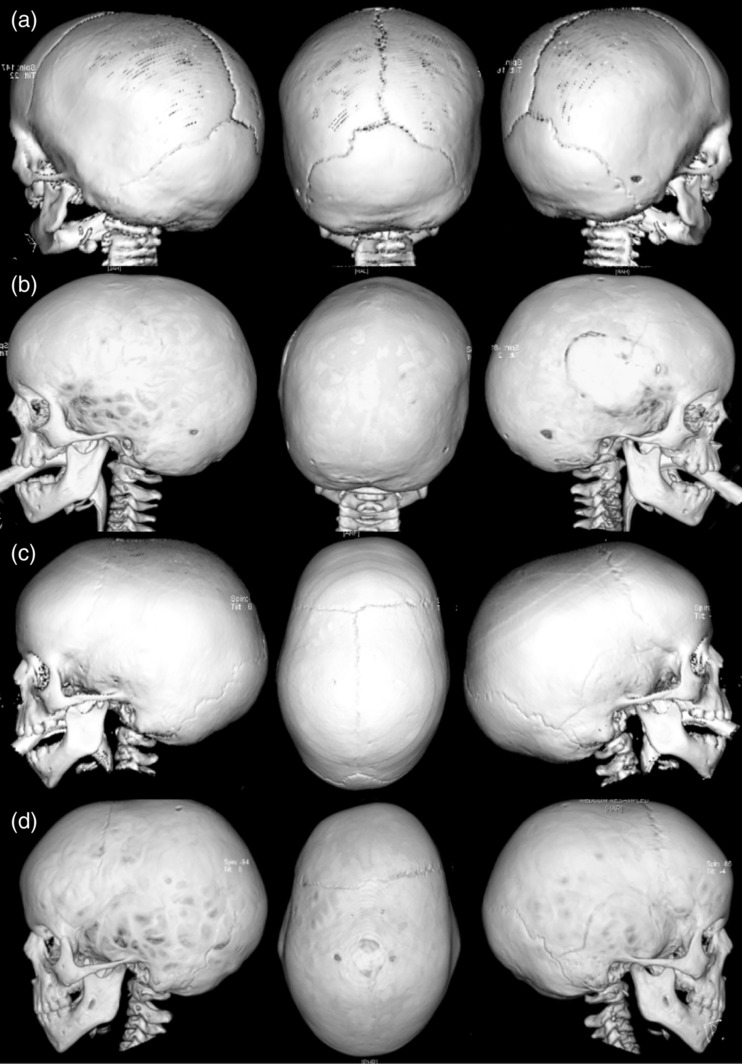
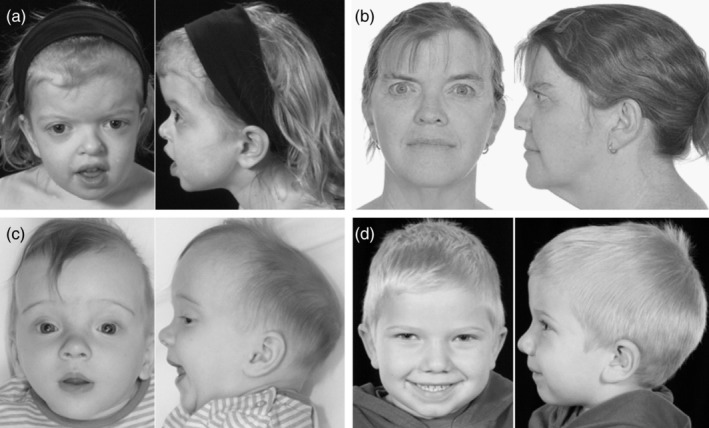
Mutations in the ERF gene, coding for ETS2 repressor factor, a member of the ETS family of transcription factors cause a recently recognized syndromic form of craniosynostosis (CRS4) with facial dysmorphism, Chiari-1 malformation, speech and language delay, and learning difficulties and/or behavioral problems. The overall prevalence of ERF mutations in patients with syndromic craniosynostosis is around 2%, and 0.7% in clinically nonsyndromic craniosynostosis. Here, we present findings from 16 unrelated probands with ERF-related craniosynostosis, with additional data from 20 family members sharing the mutations. Most of the probands exhibited multisutural (including pan-) synostosis but a pattern involving the sagittal and lambdoid sutures (Mercedes-Benz pattern) predominated. Importantly the craniosynostosis was often postnatal in onset, insidious and progressive with subtle effects on head morphology resulting in a median age at presentation of 42 months among the probands and, in some instances, permanent visual impairment due to unsuspected raised intracranial pressure (ICP). Facial dysmorphism (exhibited by all of the probands and many of the affected relatives) took the form of orbital hypertelorism, mild exorbitism and malar hypoplasia resembling Crouzon syndrome but, importantly, a Class I occlusal relationship. Speech delay, poor gross and/or fine motor control, hyperactivity and poor concentration were common. Cranial vault surgery for raised ICP and/or Chiari-1 malformation was expected when multisutural synostosis was observed. Variable expressivity and nonpenetrance among genetically affected relatives was encountered. These observations form the most complete phenotypic and developmental profile of this recently identified craniosynostosis syndrome yet described and have important implications for surgical intervention and follow-up.
Keywords: Chiari-1 malformation; ERF; craniosynostosis; facial dysmorphism; intracranial pressure; phenotype.
© 2019 The Authors. American Journal of Medical Genetics Part A published by Wiley Periodicals, Inc.
Figures
References
-
- Balasubramanian, M. , Lord, H. , Levesque, S. , Guturu, H. , Thuriot, F. , Sillon, G. , … Chitayat, D. (2017). Chitayat syndrome: Hyperphalangism, characteristic facies, hallux valgus and bronchomalacia results from a recurrent c.266a>g p.(Tyr89Cys) variant in the ERF gene. Journal of Medical Genetics, 54(3), 157–165. 10.1136/jmedgenet-2016-104143 - DOI - PubMed
-
- Goos, J. A. C. , Fenwick, A. L. , Swagemakers, S. M. A. , McGowan, S. J. , Knight, S. J. L. , Twigg, S. R. F. , … van den Ouweland, A. M. W. (2016). Identification of intragenic exon deletions and duplication of TCF12 by whole genome or targeted sequencing as a cause of TCF12‐related craniosynostosis. Human Mutation, 37(8), 732–736. 10.1002/humu.23010 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
