

Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology
- PMID: 30758993
- PMCID: PMC6580160
- DOI: 10.1152/ajpcell.00523.2018
Posttranslational modifications of mitochondrial fission and fusion proteins in cardiac physiology and pathophysiology
Abstract
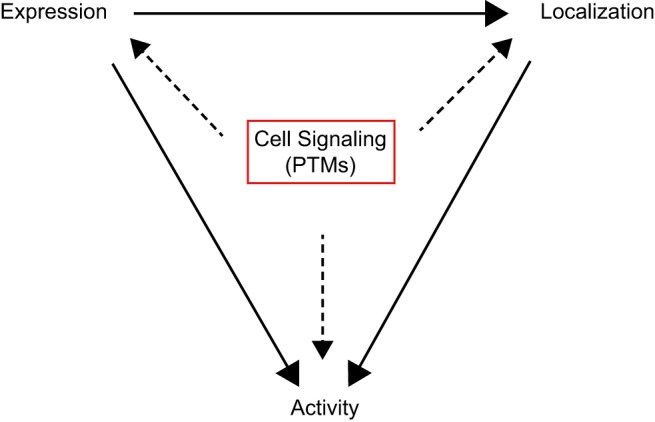
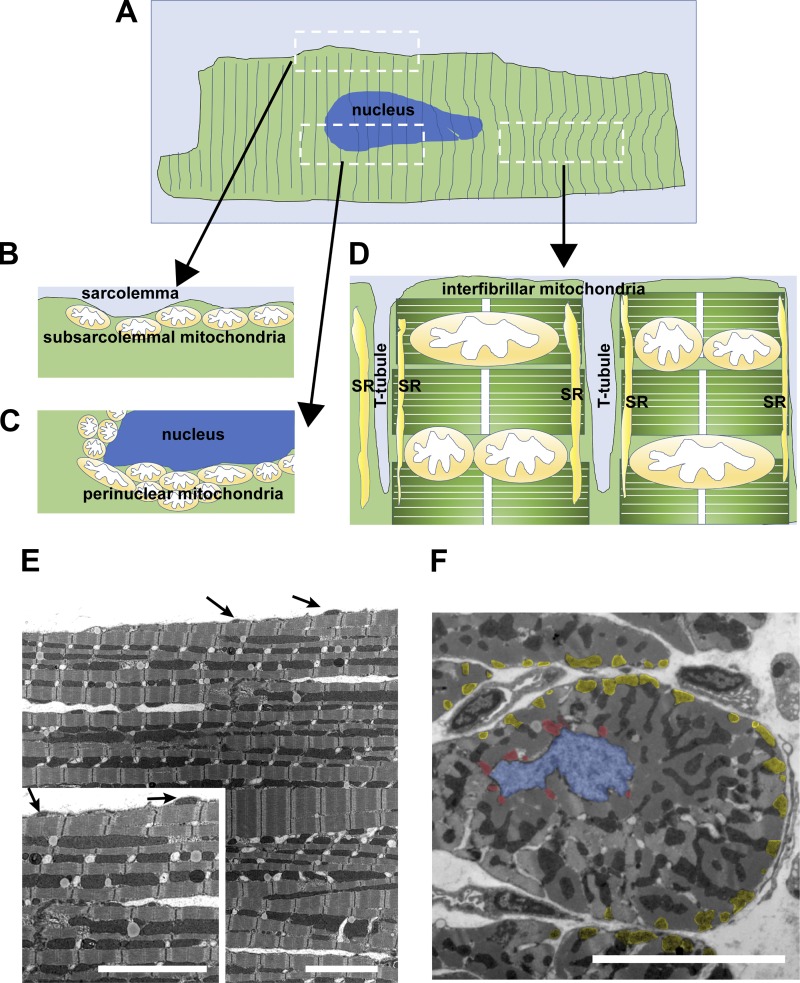
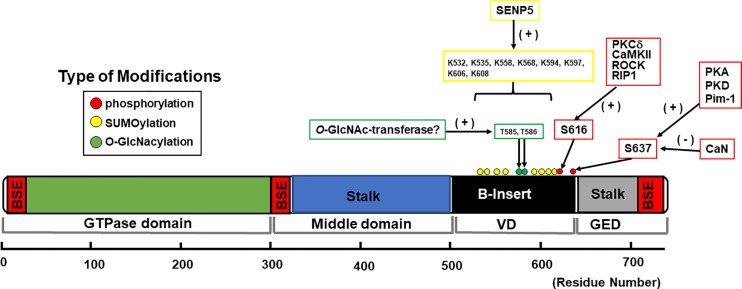
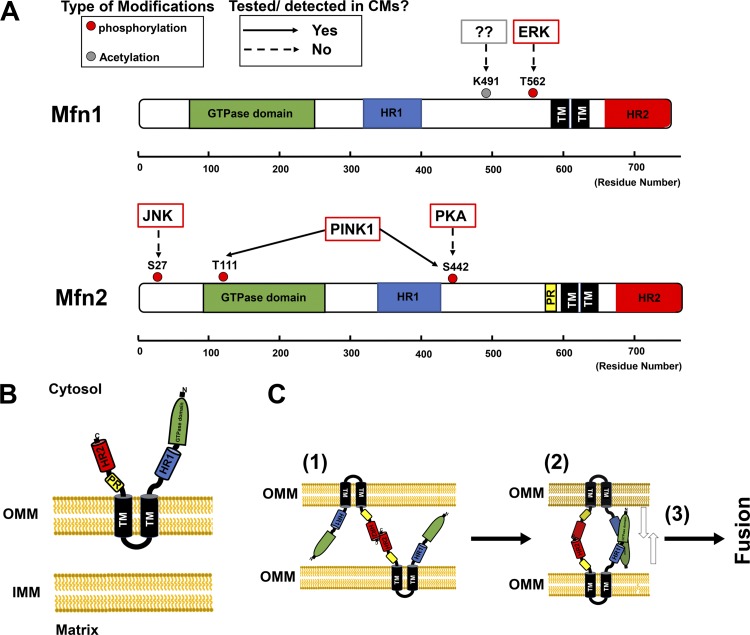
Mitochondrial fragmentation frequently occurs in chronic pathological conditions as seen in various human diseases. In fact, abnormal mitochondrial morphology and mitochondrial dysfunction are hallmarks of heart failure (HF) in both human patients and HF animal models. A link between mitochondrial fragmentation and cardiac pathologies has been widely proposed, but the physiological relevance of mitochondrial fission and fusion in the heart is still unclear. Recent studies have increasingly shown that posttranslational modifications (PTMs) of fission and fusion proteins are capable of directly modulating the stability, localization, and/or activity of these proteins. These PTMs include phosphorylation, acetylation, ubiquitination, conjugation of small ubiquitin-like modifier proteins, O-linked-N-acetyl-glucosamine glycosylation, and proteolysis. Thus, understanding the PTMs of fission and fusion proteins may allow us to understand the complexities that determine the balance of mitochondrial fission and fusion as well as mitochondrial function in various cell types and organs including cardiomyocytes and the heart. In this review, we summarize present knowledge regarding the function and regulation of mitochondrial fission and fusion in cardiomyocytes, specifically focusing on the PTMs of each mitochondrial fission/fusion protein. We also discuss the molecular mechanisms underlying abnormal mitochondrial morphology in HF and their contributions to the development of cardiac diseases, highlighting the crucial roles of PTMs of mitochondrial fission and fusion proteins. Finally, we discuss the future potential of manipulating PTMs of fission and fusion proteins as a therapeutic strategy for preventing and/or treating HF.
Keywords: DLP1; Drp1; Mfn; OPA1; mitophagy.
Conflict of interest statement
No conflicts of interest, financial or otherwise, are declared by the authors.
Figures
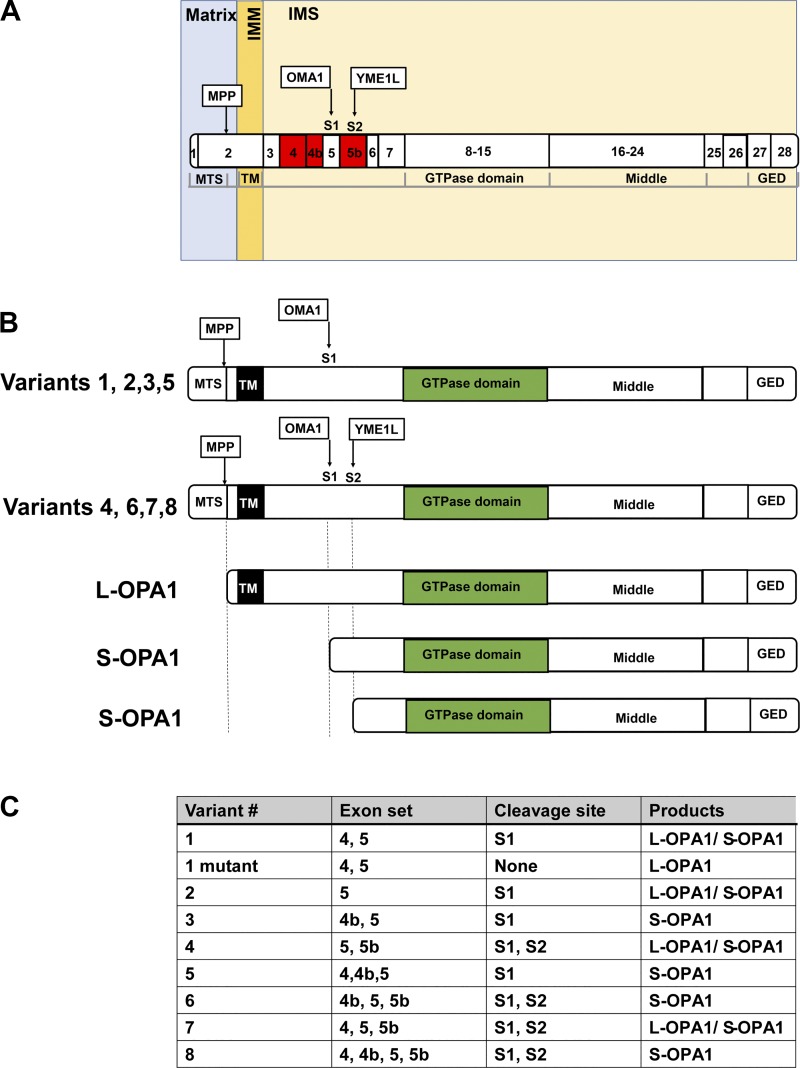
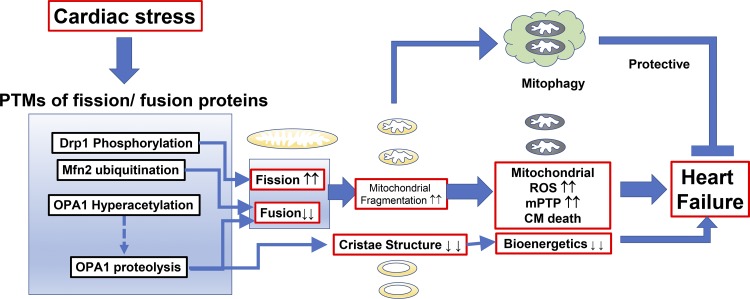
References
-
- Acin-Perez R, Lechuga-Vieco AV, Del Mar Muñoz M, Nieto-Arellano R, Torroja C, Sánchez-Cabo F, Jiménez C, González-Guerra A, Carrascoso I, Benincá C, Quiros PM, López-Otín C, Castellano JM, Ruíz-Cabello J, Jiménez-Borreguero LJ, Enríquez JA. Ablation of the stress protease OMA1 protects against heart failure in mice. Sci Transl Med 10: eaan4935, 2018. doi: 10.1126/scitranslmed.aan4935. - DOI - PubMed
-
- Alexander C, Votruba M, Pesch UE, Thiselton DL, Mayer S, Moore A, Rodriguez M, Kellner U, Leo-Kottler B, Auburger G, Bhattacharya SS, Wissinger B. OPA1, encoding a dynamin-related GTPase, is mutated in autosomal dominant optic atrophy linked to chromosome 3q28. Nat Genet 26: 211–215, 2000. doi: 10.1038/79944. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous