

PyMINEr Finds Gene and Autocrine-Paracrine Networks from Human Islet scRNA-Seq
- PMID: 30759402
- PMCID: PMC6394844
- DOI: 10.1016/j.celrep.2019.01.063
PyMINEr Finds Gene and Autocrine-Paracrine Networks from Human Islet scRNA-Seq
Abstract
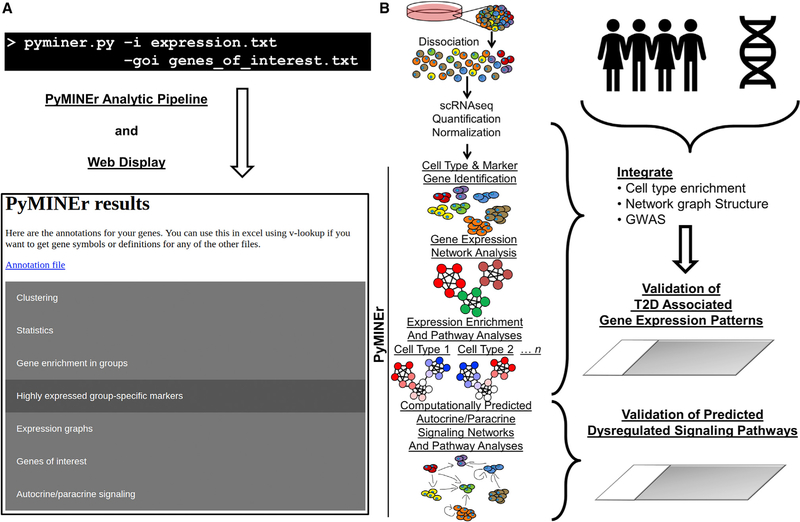
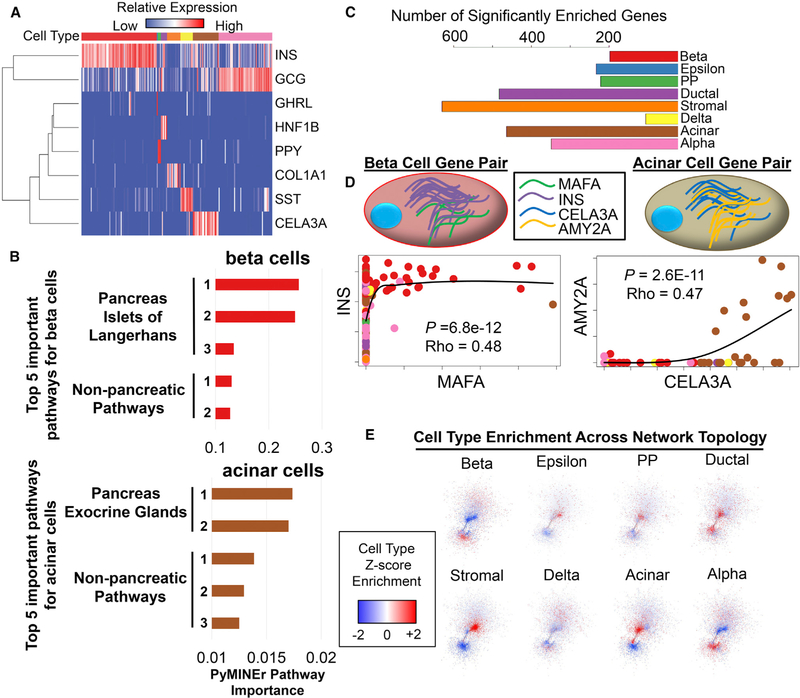
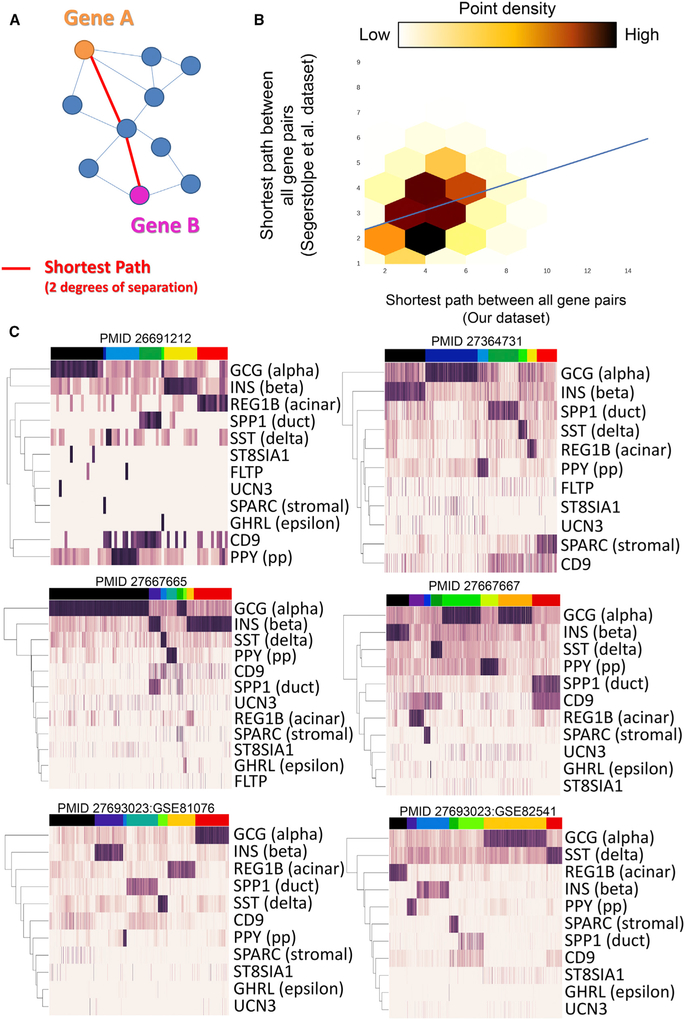
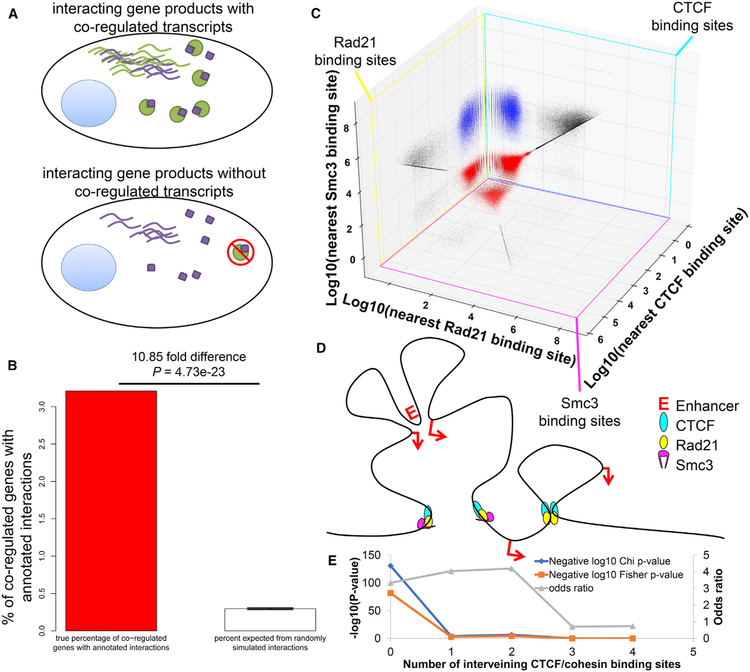
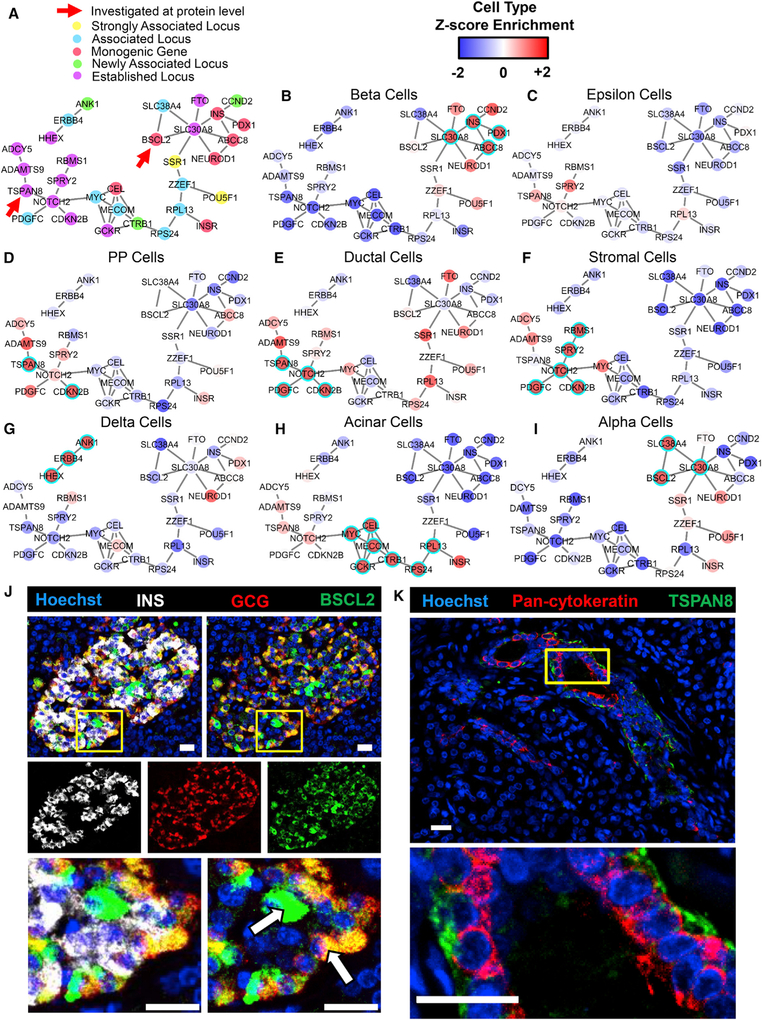
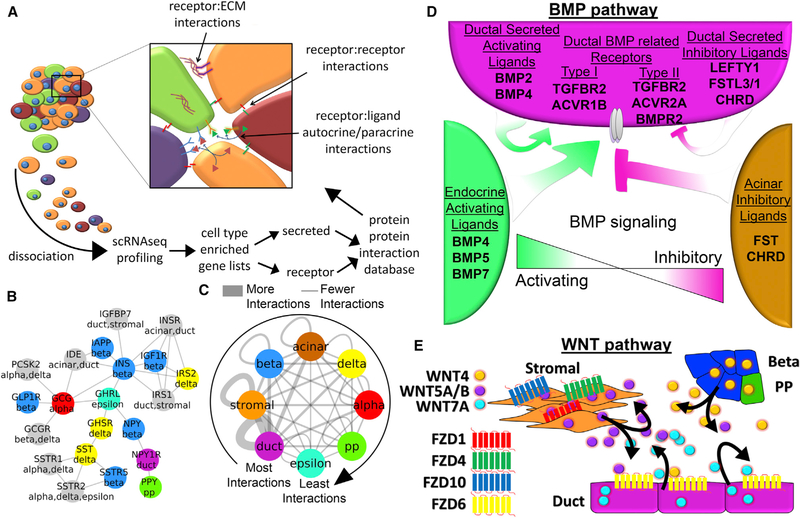
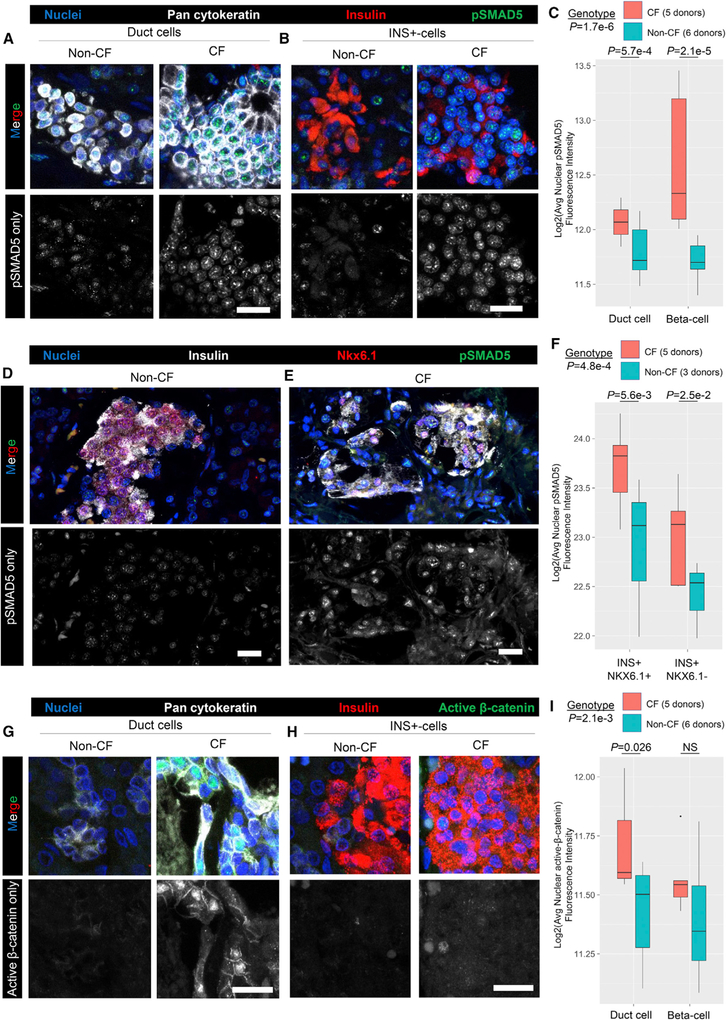
Toolsets available for in-depth analysis of scRNA-seq datasets by biologists with little informatics experience is limited. Here, we describe an informatics tool (PyMINEr) that fully automates cell type identification, cell type-specific pathway analyses, graph theory-based analysis of gene regulation, and detection of autocrine-paracrine signaling networks in silico. We applied PyMINEr to interrogate human pancreatic islet scRNA-seq datasets and discovered several features of co-expression graphs, including concordance of scRNA-seq-graph structure with both protein-protein interactions and 3D genomic architecture, association of high-connectivity and low-expression genes with cell type enrichment, and potential for the graph structure to clarify potential etiologies of enigmatic disease-associated variants. We further created a consensus co-expression network and autocrine-paracrine signaling networks within and across islet cell types from seven datasets. PyMINEr correctly identified changes in BMP-WNT signaling associated with cystic fibrosis pancreatic acinar cell loss. This proof-of-principle study demonstrates that the PyMINEr framework will be a valuable resource for scRNA-seq analyses.
Keywords: BMP; PyMINEr; WNT; autocrine-paracrine; cell type identification; cystic fibrosis; networks; pancreatic islets; single-cell RNA-seq; systems biology.
Copyright © 2019 The Author(s). Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no conflicts of interest.
Figures
References
-
- Arthur D, and Vassilvitskii S (2007). k-means++: the advantages of careful seeding In Proceedings of the Eighteenth Annual ACM-SIAM Symposium on Discrete Algorithms (Society for Industrial and Applied Mathematics), pp. 1027–1035.
-
- Bader E, Migliorini A, Gegg M, Moruzzi N, Gerdes J, Roscioni SS, Bakhti M, Brandl E, Irmler M, Beckers J, et al. (2016). Identification of proliferative and mature b-cells in the islets of Langerhans. Nature 535, 430–434. - PubMed
-
- Behfar A, Zingman LV, Hodgson DM, Rauzier J-M, Kane GC, Terzic A, and Pucéat M (2002). Stem cell differentiation requires a paracrine pathway in the heart. FASEB J 16, 1558–1566. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
