

A mole rat's gut microbiota suggests selective influence of diet on microbial niche space and evolution
- PMID: 30760029
- PMCID: PMC6547004
- DOI: 10.1177/1535370219828703
A mole rat's gut microbiota suggests selective influence of diet on microbial niche space and evolution
Abstract
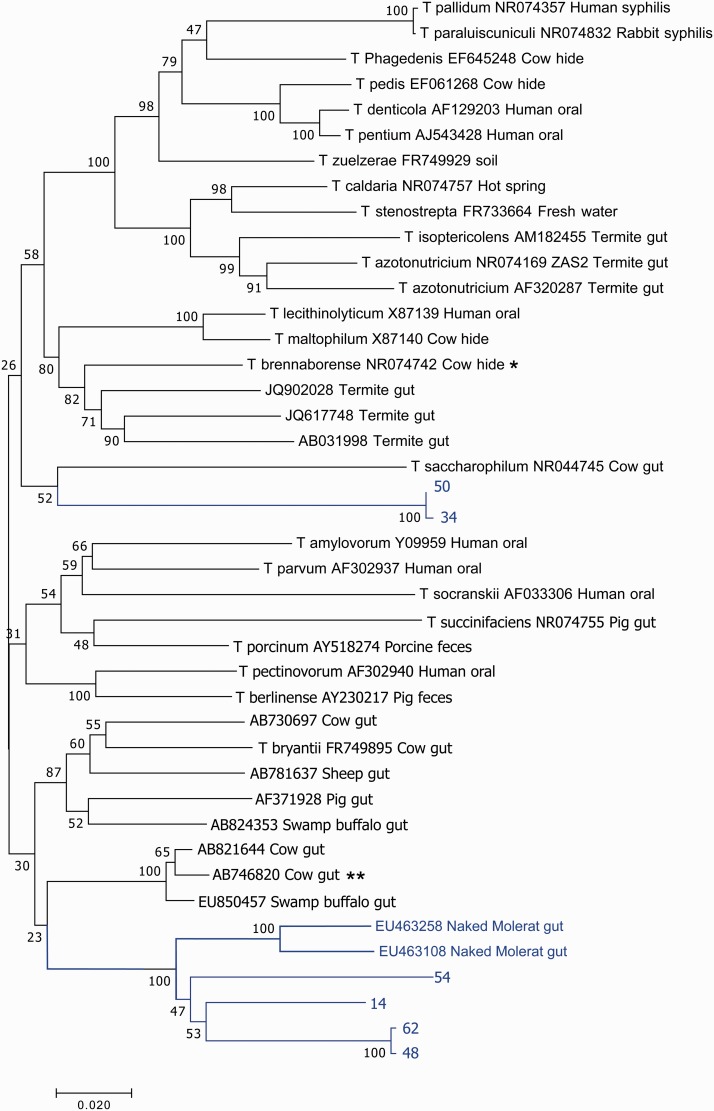
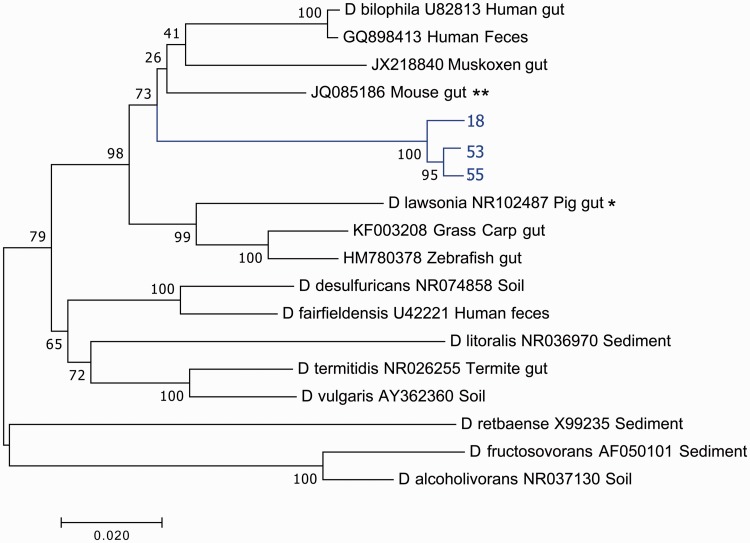
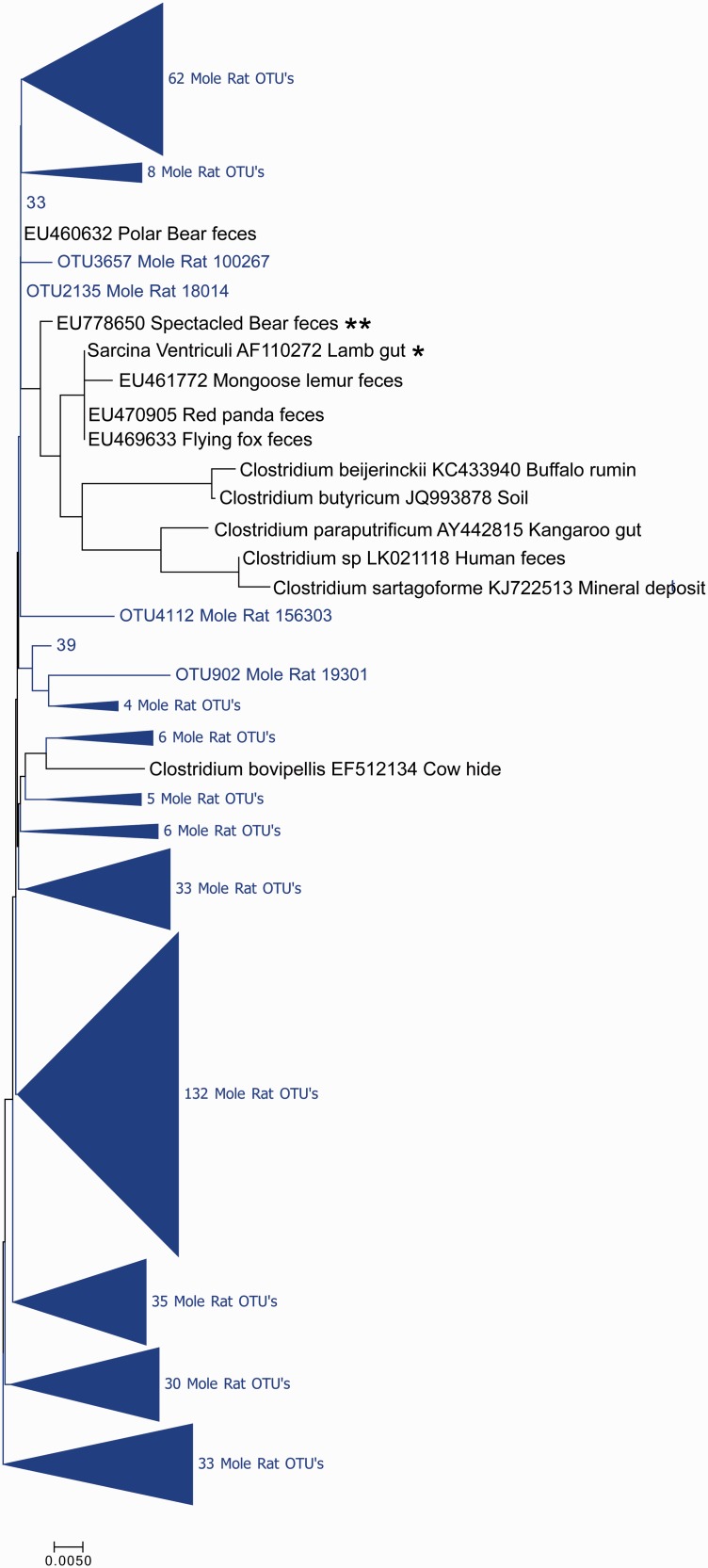
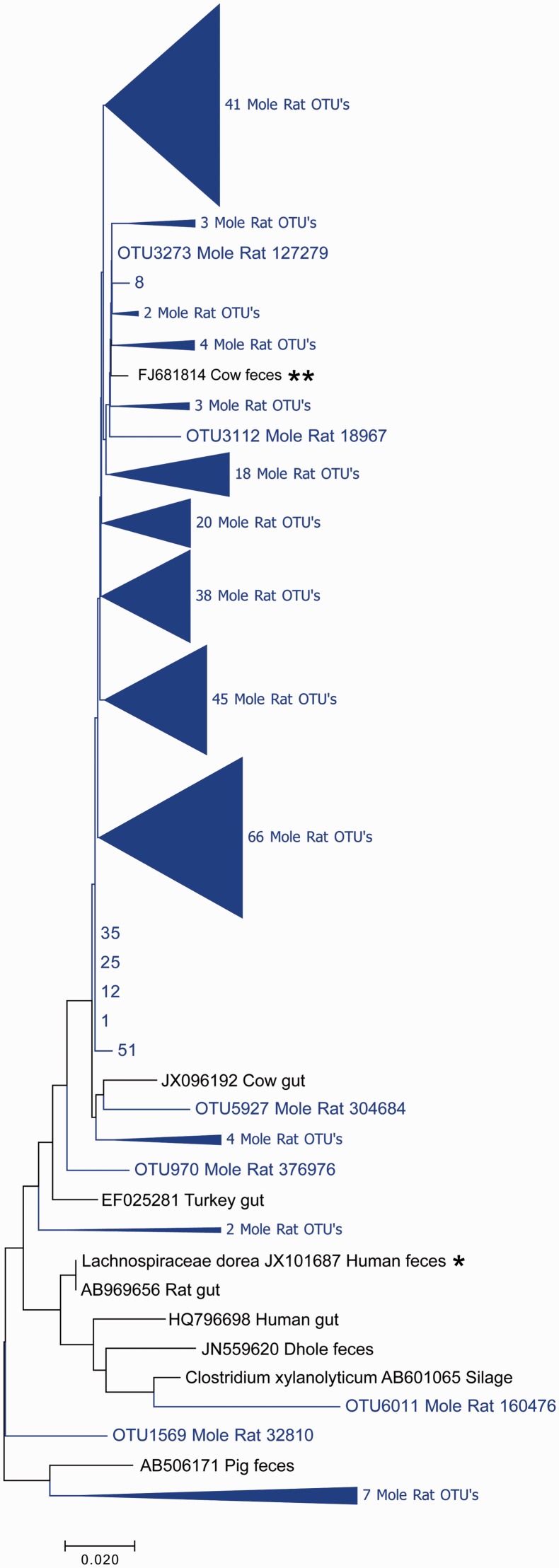
The composition of the microbiota is of critical importance for health and disease, and is receiving increased scientific and medical scrutiny. Of particular interest is the role of changing diets as a function of agriculture and, perhaps to an even greater extent, modern food processing. To probe the connection between diet and the gut's microbial community, the microbiota from a mole rat, a rodent with a relatively unusual diet, was analyzed in detail, and the microbes found were compared with previously identified organisms. The results show evidence of an adaptive radiation of some microbial clades, but relative stability in others. This suggests that the microbiota, like the genome, carries with it housekeeping components as well as other components which can evolve rapidly when the environment changes. This study provides a very broad view of the niche space in the gut and how factors such as diet might influence that niche space.
Keywords: Diet; evolution; fiber; microbiota; mole rat; niche.
Figures




References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources