

The US11 Gene of Herpes Simplex Virus 1 Promotes Neuroinvasion and Periocular Replication following Corneal Infection
- PMID: 30760571
- PMCID: PMC6475787
- DOI: 10.1128/JVI.02246-18
The US11 Gene of Herpes Simplex Virus 1 Promotes Neuroinvasion and Periocular Replication following Corneal Infection
Abstract
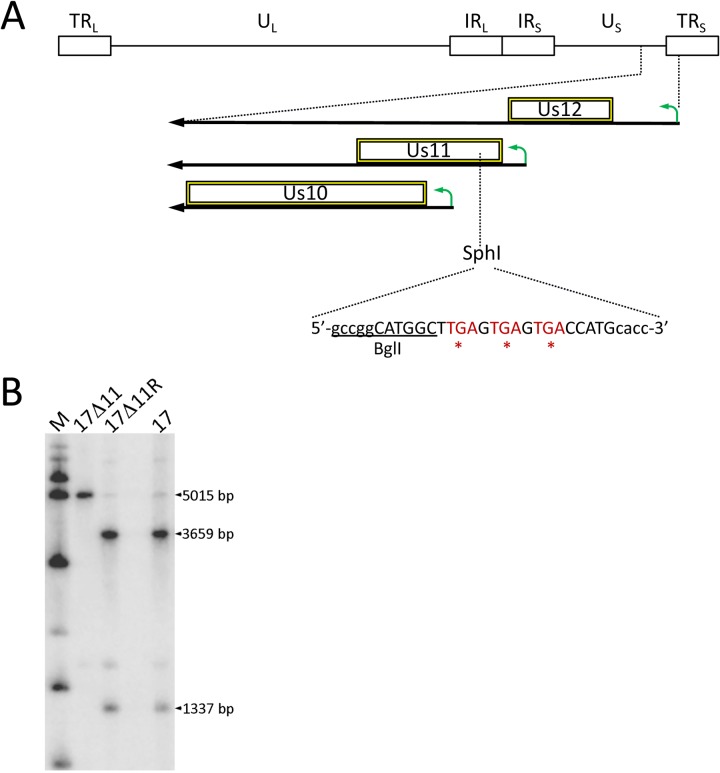
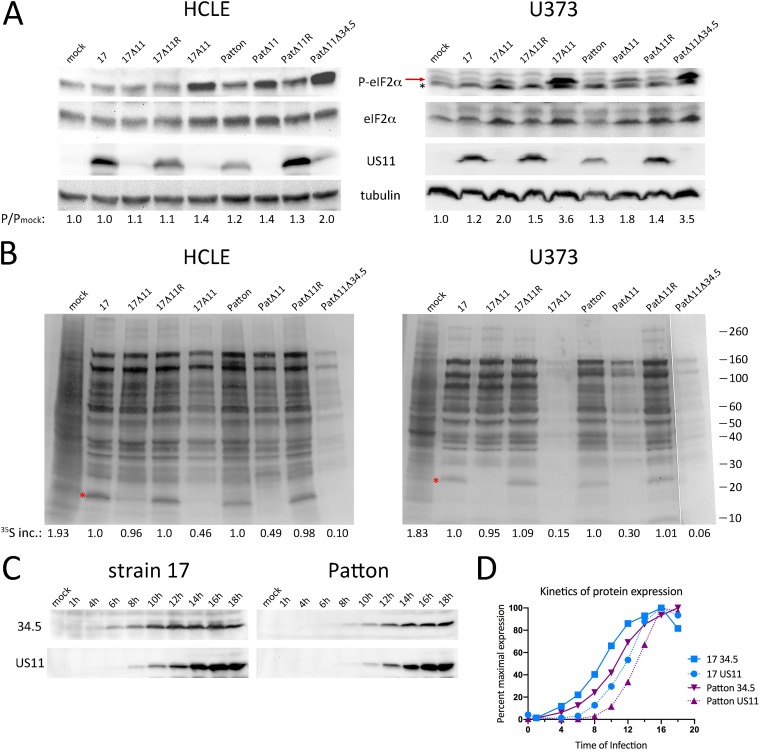
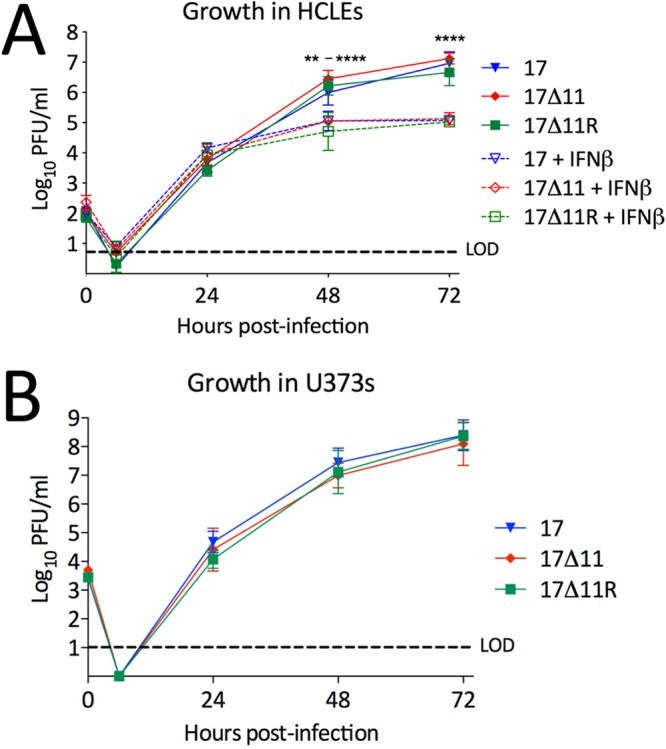
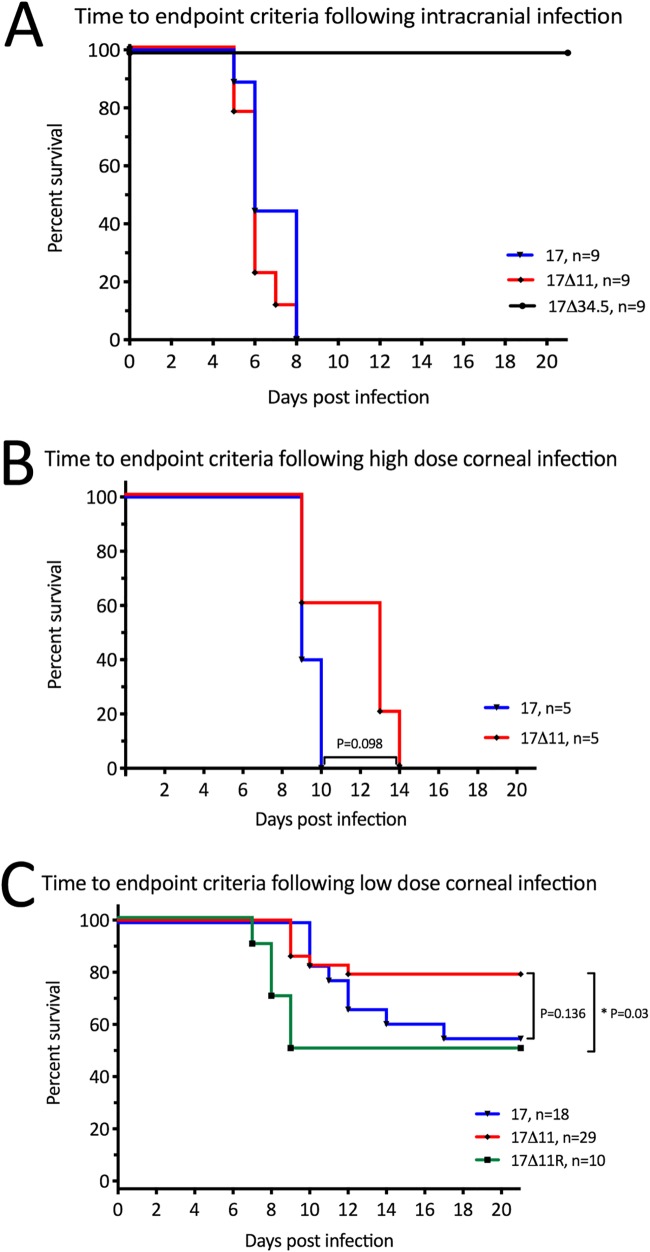
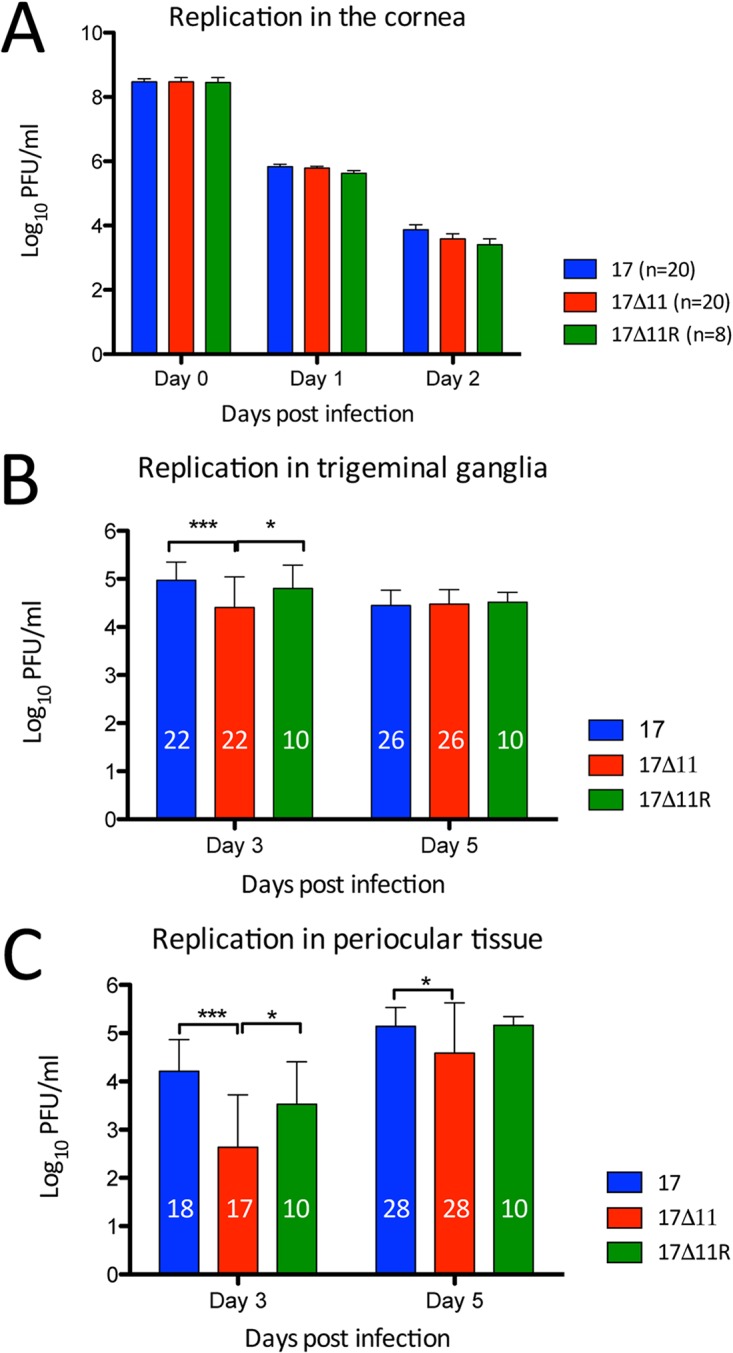
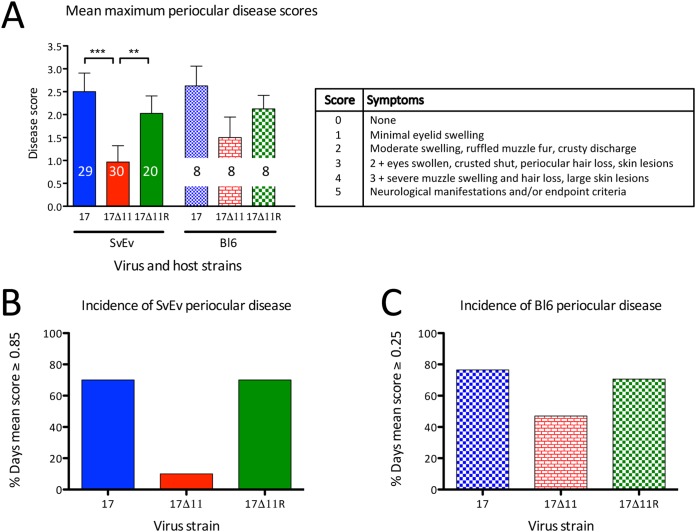
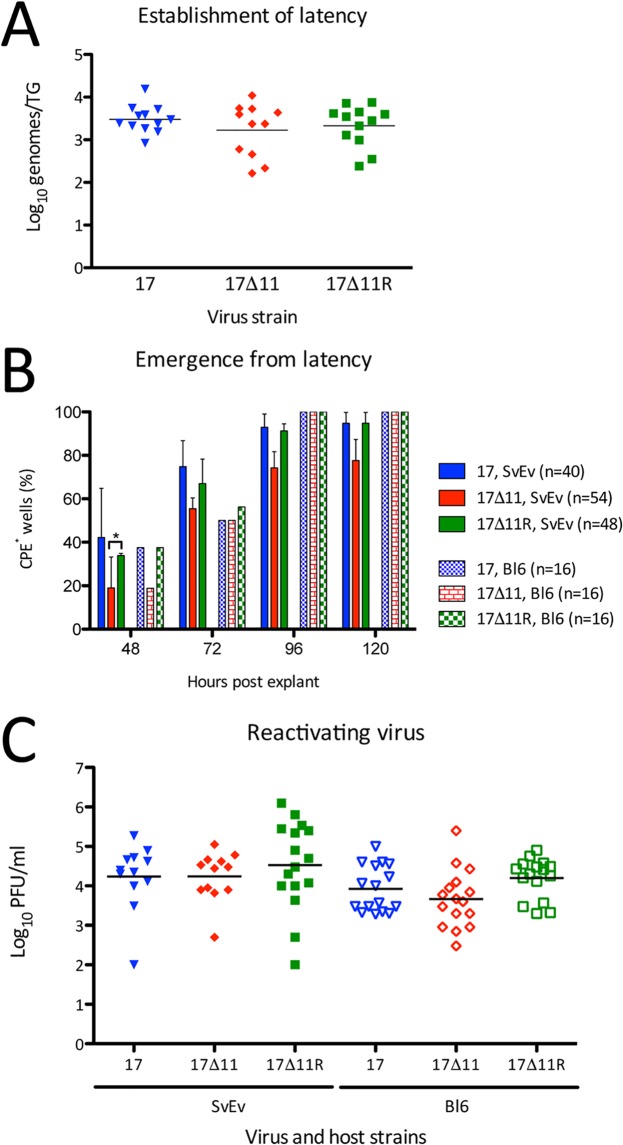
Herpes simplex virus 1 (HSV-1) cycles between phases of latency in sensory neurons and replication in mucosal sites. HSV-1 encodes two key proteins that antagonize the shutdown of host translation, US11 through preventing PKR activation and ICP34.5 through mediating dephosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α). While profound attenuation of ICP34.5 deletion mutants has been repeatedly demonstrated, a role for US11 in HSV-1 pathogenesis remains unclear. We therefore generated an HSV-1 strain 17 US11-null virus and examined its properties in vitro and in vivo In U373 glioblastoma cells, US11 cooperated with ICP34.5 to prevent eIF2α phosphorylation late in infection. However, the effect was muted in human corneal epithelial cells (HCLEs), which did not accumulate phosphorylated eIF2α unless both US11 and ICP34.5 were absent. Low levels of phosphorylated eIF2α correlated with continued protein synthesis and with the ability of virus lacking US11 to overcome antiviral immunity in HCLE and U373 cells. Neurovirulence following intracerebral inoculation of mice was not affected by the deletion of US11. In contrast, the time to endpoint criteria following corneal infection was greater for the US11-null virus than for the wild-type virus. Replication in trigeminal ganglia and periocular tissue was promoted by US11, as was periocular disease. The establishment of latency and the frequency of virus reactivation from trigeminal ganglia were unaffected by US11 deletion, although emergence of the US11-null virus occurred with slowed kinetics. Considered together, the data indicate that US11 facilitates the countering of antiviral response of infected cells and promotes the efficient emergence of virus following reactivation.IMPORTANCE Alphaherpesviruses are ubiquitous DNA viruses and include the human pathogens herpes simplex virus 1 (HSV-1) and HSV-2 and are significant causes of ulcerative mucosal sores, infectious blindness, encephalitis, and devastating neonatal disease. Successful primary infection and persistent coexistence with host immune defenses are dependent on the ability of these viruses to counter the antiviral response. HSV-1 and HSV-2 and other primate viruses within the Simplexvirus genus encode US11, an immune antagonist that promotes virus production by preventing shutdown of protein translation. Here we investigated the impact of US11 deletion on HSV-1 growth in vitro and pathogenesis in vivo This work supports a role for US11 in pathogenesis and emergence from latency, elucidating immunomodulation by this medically important cohort of viruses.
Keywords: herpes simplex virus; innate immunity; pathogenesis.
Copyright © 2019 American Society for Microbiology.
Figures
Similar articles
-
In vivo replication of an ICP34.5 second-site suppressor mutant following corneal infection correlates with in vitro regulation of eIF2 alpha phosphorylation.J Virol. 2003 Apr;77(8):4626-34. doi: 10.1128/jvi.77.8.4626-4634.2003. J Virol. 2003. PMID: 12663769 Free PMC article.
-
The herpes simplex virus US11 protein effectively compensates for the gamma1(34.5) gene if present before activation of protein kinase R by precluding its phosphorylation and that of the alpha subunit of eukaryotic translation initiation factor 2.J Virol. 1998 Nov;72(11):8620-6. doi: 10.1128/JVI.72.11.8620-8626.1998. J Virol. 1998. PMID: 9765401 Free PMC article.
-
The herpes simplex virus 1 Us11 protein inhibits autophagy through its interaction with the protein kinase PKR.J Virol. 2013 Jan;87(2):859-71. doi: 10.1128/JVI.01158-12. Epub 2012 Oct 31. J Virol. 2013. PMID: 23115300 Free PMC article.
-
Neutralizing innate host defenses to control viral translation in HSV-1 infected cells.Int Rev Immunol. 2004 Jan-Apr;23(1-2):199-220. doi: 10.1080/08830180490265600. Int Rev Immunol. 2004. PMID: 14690861 Review.
-
Control of HSV-1 latency in human trigeminal ganglia--current overview.J Neurovirol. 2011 Dec;17(6):518-27. doi: 10.1007/s13365-011-0063-0. Epub 2011 Dec 3. J Neurovirol. 2011. PMID: 22139603 Review.
Cited by
-
Advanced progress in the genetic modification of the oncolytic HSV-1 virus.Front Oncol. 2025 Jan 21;14:1525940. doi: 10.3389/fonc.2024.1525940. eCollection 2024. Front Oncol. 2025. PMID: 39906660 Free PMC article. Review.
-
An Intrinsic Host Defense against HSV-1 Relies on the Activation of Xenophagy with the Active Clearance of Autophagic Receptors.Cells. 2024 Jul 26;13(15):1256. doi: 10.3390/cells13151256. Cells. 2024. PMID: 39120287 Free PMC article.
-
Post-translational modifications as a key mechanism for herpes simplex virus type I evasion of host innate immunity.Front Microbiol. 2025 Feb 11;16:1543676. doi: 10.3389/fmicb.2025.1543676. eCollection 2025. Front Microbiol. 2025. PMID: 40008039 Free PMC article. Review.
-
Recent Advances in the Study of Alphaherpesvirus Latency and Reactivation: Novel Guidance for the Design of Herpesvirus Live Vector Vaccines.Pathogens. 2024 Sep 10;13(9):779. doi: 10.3390/pathogens13090779. Pathogens. 2024. PMID: 39338969 Free PMC article. Review.
-
"Non-Essential" Proteins of HSV-1 with Essential Roles In Vivo: A Comprehensive Review.Viruses. 2020 Dec 23;13(1):17. doi: 10.3390/v13010017. Viruses. 2020. PMID: 33374862 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
