

High frequency of shared clonotypes in human B cell receptor repertoires
- PMID: 30760926
- PMCID: PMC6949180
- DOI: 10.1038/s41586-019-0934-8
High frequency of shared clonotypes in human B cell receptor repertoires
Abstract
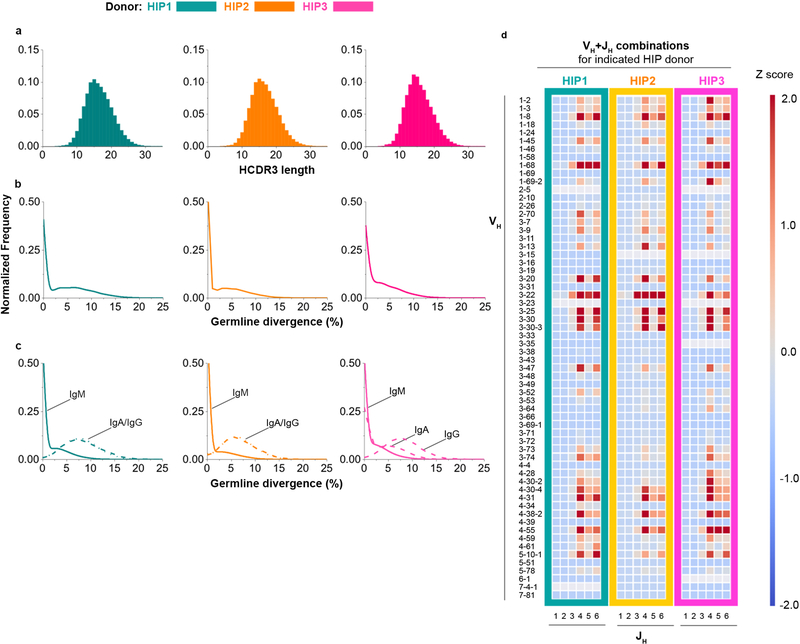
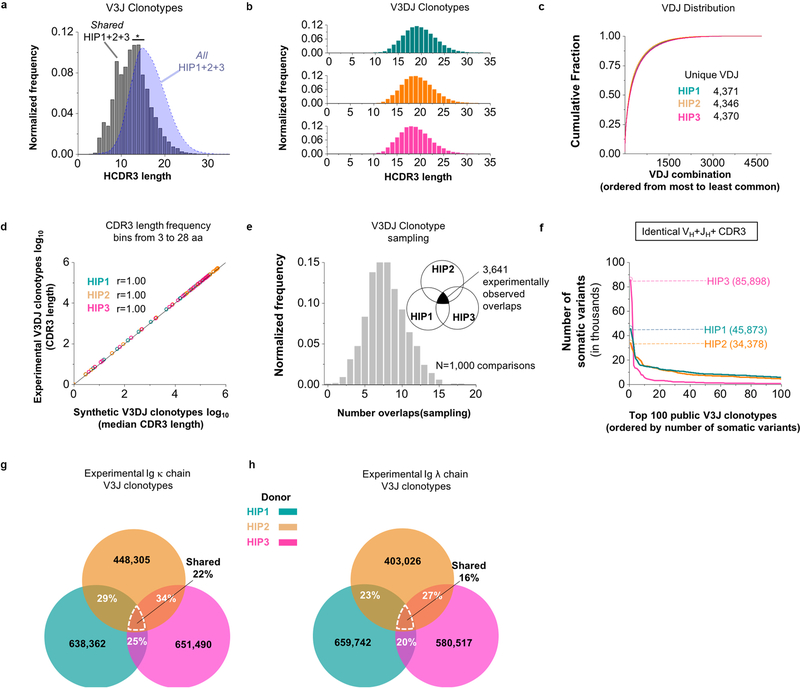
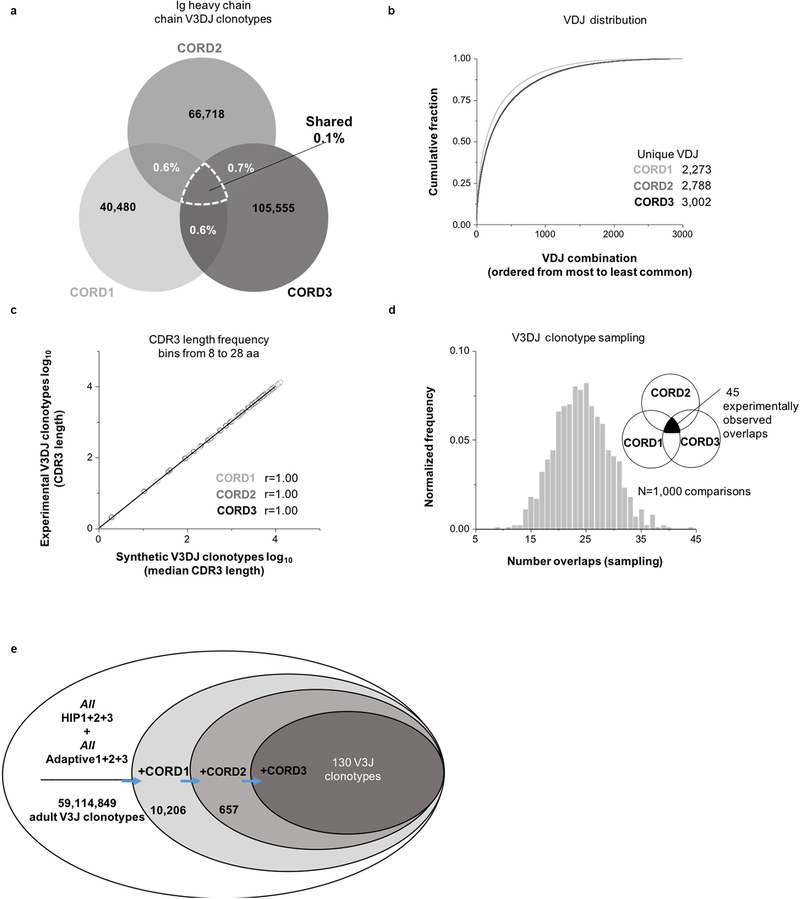
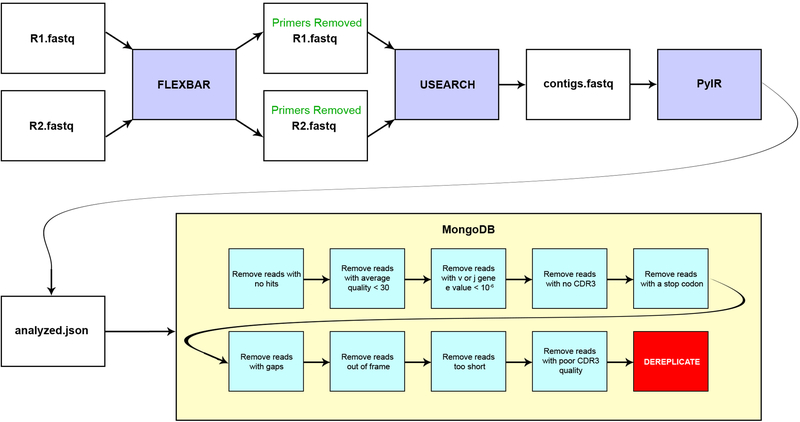
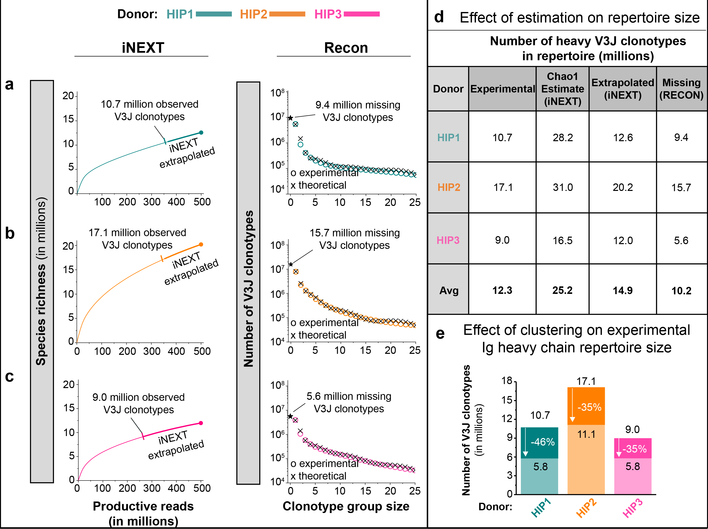
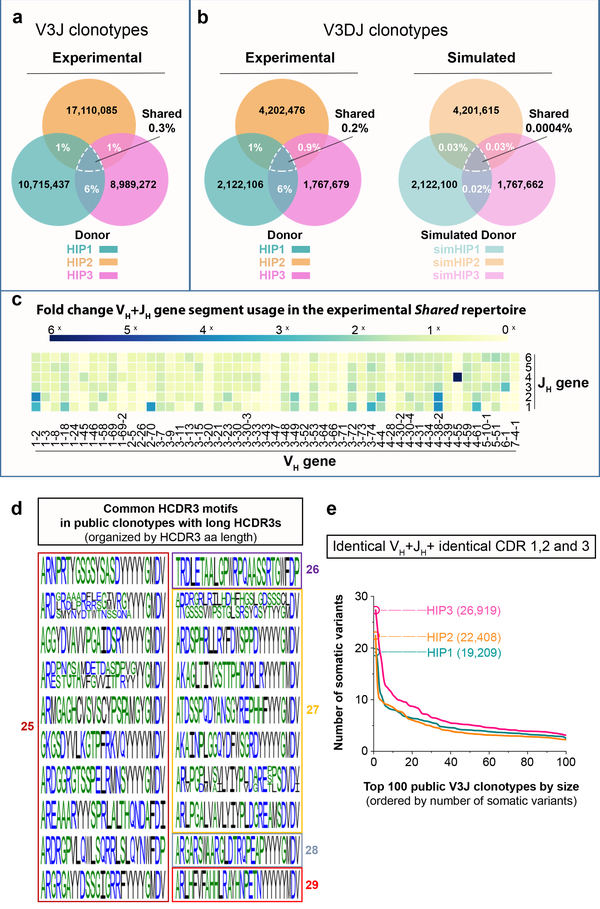
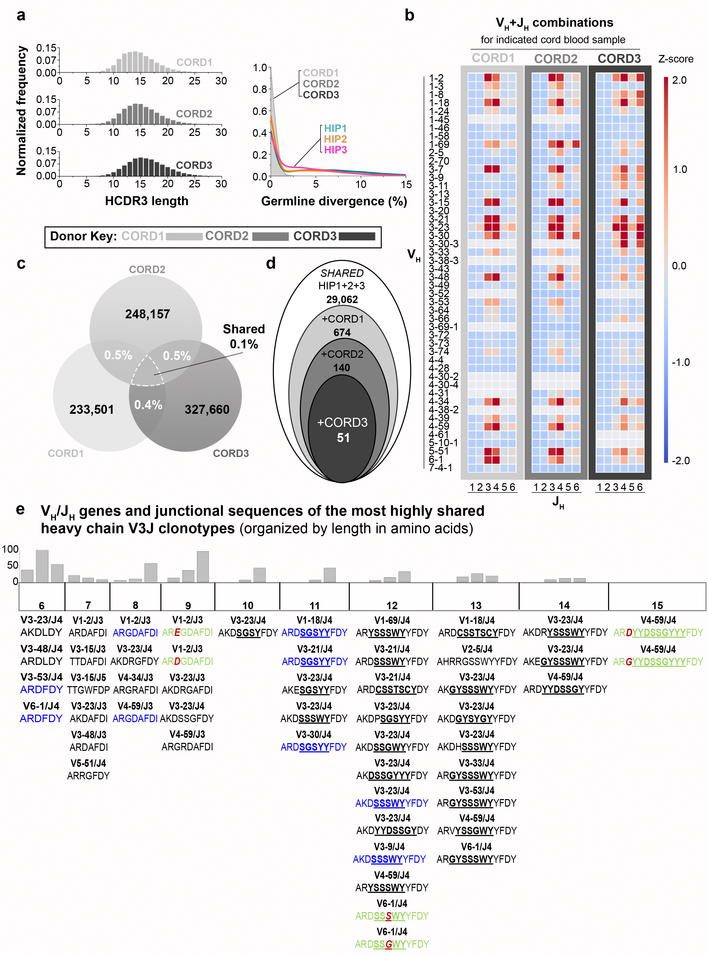
The human genome contains approximately 20 thousand protein-coding genes1, but the size of the collection of antigen receptors of the adaptive immune system that is generated by the recombination of gene segments with non-templated junctional additions (on B cells) is unknown-although it is certainly orders of magnitude larger. It has not been established whether individuals possess unique (or private) repertoires or substantial components of shared (or public) repertoires. Here we sequence recombined and expressed B cell receptor genes in several individuals to determine the size of their B cell receptor repertoires, and the extent to which these are shared between individuals. Our experiments revealed that the circulating repertoire of each individual contained between 9 and 17 million B cell clonotypes. The three individuals that we studied shared many clonotypes, including between 1 and 6% of B cell heavy-chain clonotypes shared between two subjects (0.3% of clonotypes shared by all three) and 20 to 34% of λ or κ light chains shared between two subjects (16 or 22% of λ or κ light chains, respectively, were shared by all three). Some of the B cell clonotypes had thousands of clones, or somatic variants, within the clonotype lineage. Although some of these shared lineages might be driven by exposure to common antigens, previous exposure to foreign antigens was not the only force that shaped the shared repertoires, as we also identified shared clonotypes in umbilical cord blood samples and all adult repertoires. The unexpectedly high prevalence of shared clonotypes in B cell repertoires, and identification of the sequences of these shared clonotypes, should enable better understanding of the role of B cell immune repertoires in health and disease.
Figures

References
-
- The Adaptive Immune Receptor Repertoire Community of the Antibody Society <https://www.antibodysociety.org/the-airr-community/>
-
- Hsieh TC, Ma KH & Chao A iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol Evol 7, 1451–1456, (2016).
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
