

MT-ATP6 mitochondrial disease variants: Phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases
- PMID: 30763462
- PMCID: PMC6506718
- DOI: 10.1002/humu.23723
MT-ATP6 mitochondrial disease variants: Phenotypic and biochemical features analysis in 218 published cases and cohort of 14 new cases
Abstract
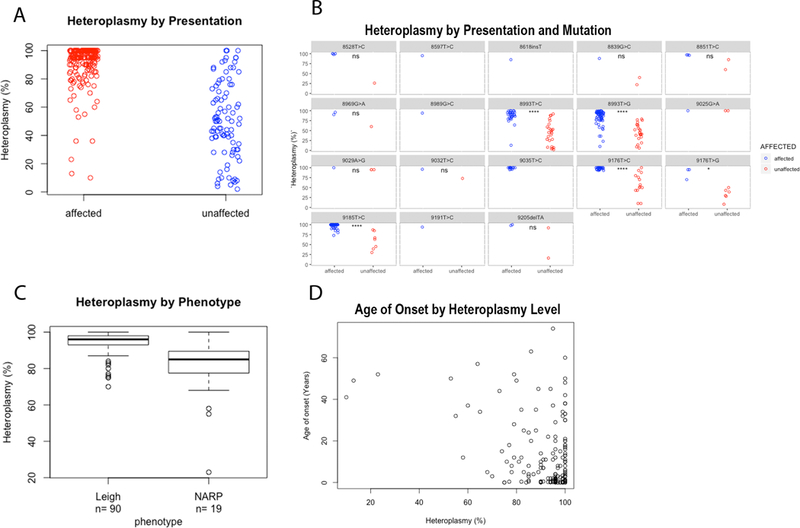
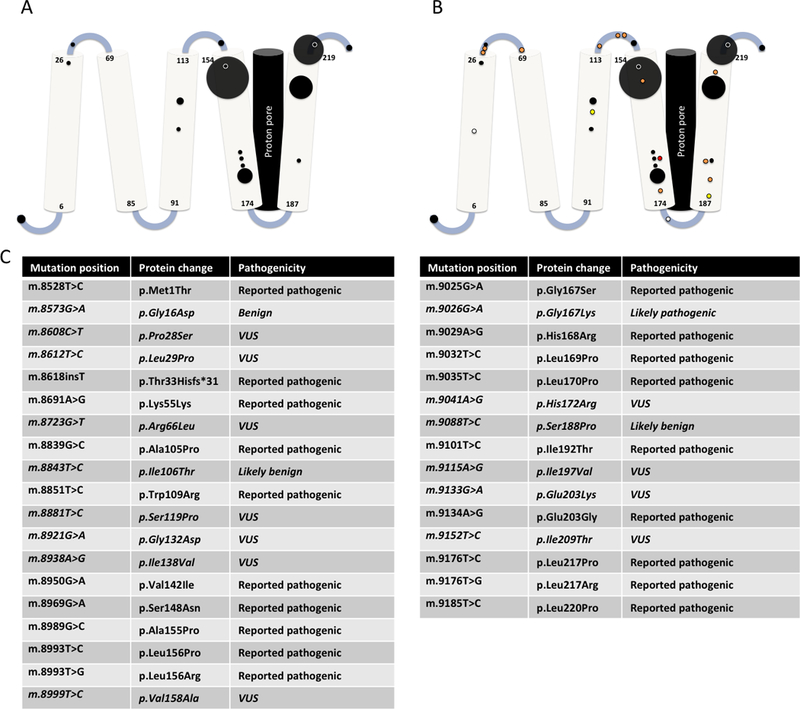
Mitochondrial complex V (CV) generates cellular energy as adenosine triphosphate (ATP). Mitochondrial disease caused by the m.8993T>G pathogenic variant in the CV subunit gene MT-ATP6 was among the first described human mitochondrial DNA diseases. Due to a lack of clinically available functional assays, validating the definitive pathogenicity of additional MT-ATP6 variants remains challenging. We reviewed all reportedMT-ATP6 disease cases ( n = 218) to date, to assess for MT-ATP6 variants, heteroplasmy levels, and inheritance correlation with clinical presentation and biochemical findings. We further describe the clinical and biochemical features of a new cohort of 14 kindreds with MT-ATP6 variants of uncertain significance. Despite extensive overlap in the heteroplasmy levels of MT-ATP6 variant carriers with and without a wide range of clinical symptoms, previously reported symptomatic subjects had significantly higher heteroplasmy load (p = 2.2 x 10-16 ). Pathogenic MT-ATP6 variants resulted in diverse biochemical features. The most common findings were reduced ATP synthesis rate, preserved ATP hydrolysis capacity, and abnormally increased mitochondrial membrane potential. However, no single biochemical feature was universally observed. Extensive heterogeneity exists among both clinical and biochemical features of distinct MT-ATP6 variants. Improved mechanistic understanding and development of consistent biochemical diagnostic analyses are needed to permit accurate pathogenicity assessment of variants of uncertain significance in MT-ATP6.
Keywords: Leigh syndrome; genotype-phenotype correlation; heteroplasmy; mitochondria; neurogenic ataxia and retinitis pigmentosa.
© 2019 Wiley Periodicals, Inc.
Figures
References
-
- Abu-Amero KK, & Bosley TM (2005). Detection of Mitochondrial Respiratory Dysfunction in Circulating Lymphocytes Using Resazurin. Arch Pathol Lab Med—Vol, 129. - PubMed
-
- Alila-Fersi O, Chamkha I, Majdoub I, Gargouri L, Mkaouar-Rebai E, Tabebi M, … Fakhfakh F (2017). Co segregation of the m.1555A>G mutation in the MT-RNR1 gene and mutations in MT-ATP6 gene in a family with dilated mitochondrial cardiomyopathy and hearing loss: A whole mitochondrial genome screening. Biochemical and Biophysical Research Communications, 484(1), 71–78. 10.1016/j.bbrc.2017.01.070 - DOI - PubMed
-
- Burrage LC, Tang S, Wang J, Donti TR, Walkiewicz M, Luchak JM, … Scaglia F (2014). Mitochondrial myopathy, lactic acidosis, and sideroblastic anemia (MLASA) plus associated with a novel de novo mutation (m.8969G>A) in the mitochondrial encoded ATP6 gene. Molecular Genetics and Metabolism, 113(3), 207–212. 10.1016/j.ymgme.2014.06.004 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U54-NS078059/NS/NINDS NIH HHS/United States
- North American Mitochondrial Disease Foundation/International
- NH/NIH HHS/United States
- United Mitochondrial Disease Foundation/International
- North American Mitochondrial Disease Consortium/International
- U54 NS078059/NS/NINDS NIH HHS/United States
- K08 DK113250/DK/NIDDK NIH HHS/United States
- U24 HD093483/HD/NICHD NIH HHS/United States
- U54-HD086984/Eunice Kennedy Shriver National Institute of Child Health and Human Development/International
- U54 HD086984/HD/NICHD NIH HHS/United States
- K08-DK113250/DK/NIDDK NIH HHS/United States
- U24-HD093483/Eunice Kennedy Shriver National Institute of Child Health and Human Development/International
LinkOut - more resources
Full Text Sources
Medical
