

Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins
- PMID: 30763537
- PMCID: PMC6411085
- DOI: 10.1016/j.chom.2019.01.019
Virome Diversity Correlates with Intestinal Microbiome Diversity in Adult Monozygotic Twins
Abstract
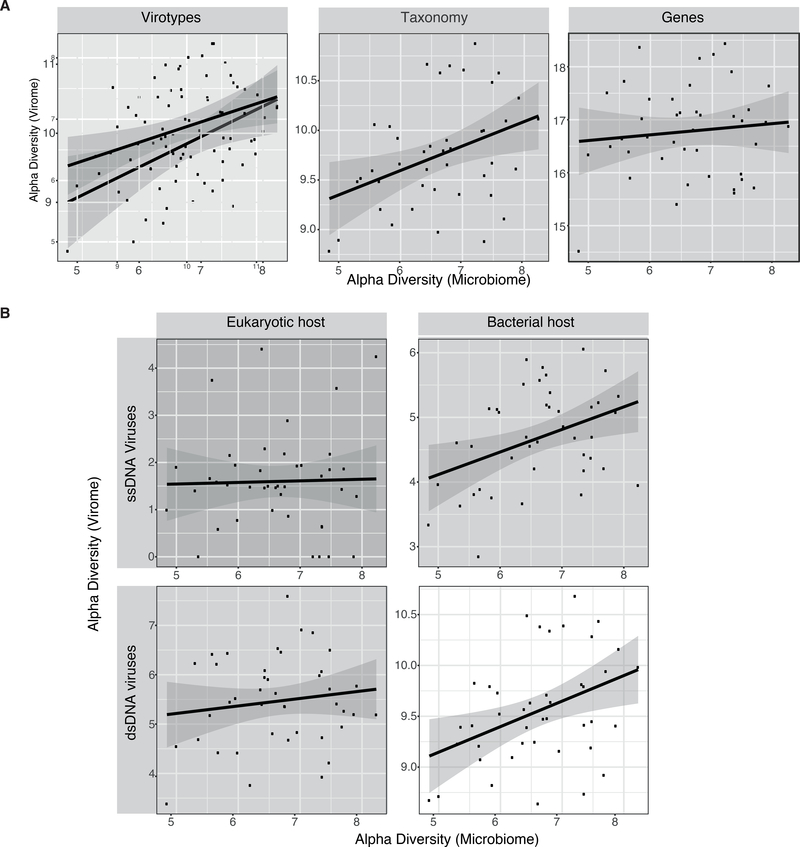
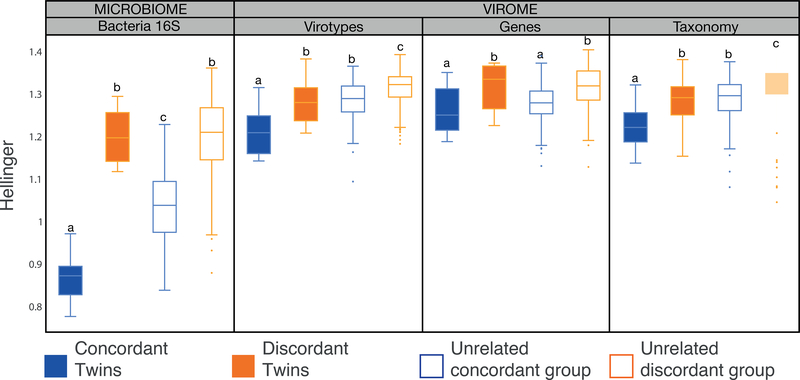
The virome is one of the most variable components of the human gut microbiome. Within twin pairs, viromes have been shown to be similar for infants, but not for adults, indicating that as twins age and their environments and microbiomes diverge, so do their viromes. The degree to which the microbiome drives the vast virome diversity is unclear. Here, we examine the relationship between microbiome and virome diversity in 21 adult monozygotic twin pairs selected for high or low microbiome concordance. Viromes derived from virus-like particles are unique to each individual, are dominated by Caudovirales and Microviridae, and exhibit a small core that includes crAssphage. Microbiome-discordant twins display more dissimilar viromes compared to microbiome-concordant twins, and the richer the microbiomes, the richer the viromes. These patterns are driven by bacteriophages, not eukaryotic viruses. Collectively, these observations support a strong role of the microbiome in patterning for the virome.
Keywords: concordant and discordant monozygotic twins; human gut microbiome; human gut virome.
Copyright © 2019 Elsevier Inc. All rights reserved.
Conflict of interest statement
DECLARATION OF INTERESTS
The authors declare no competing interests.
Figures
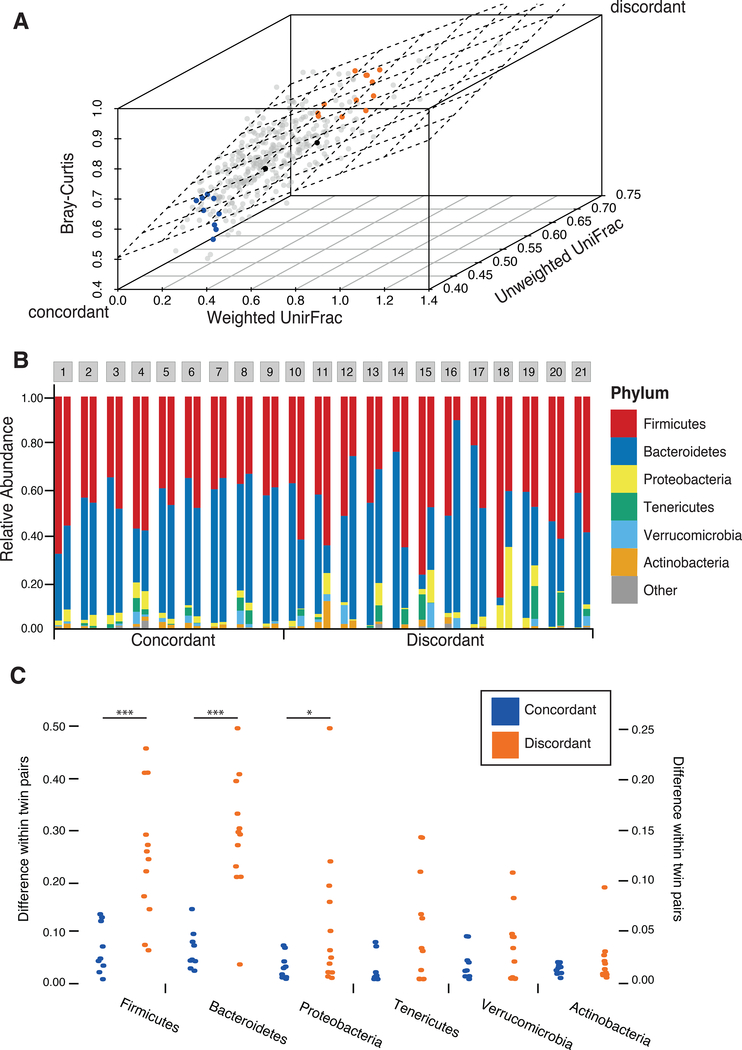
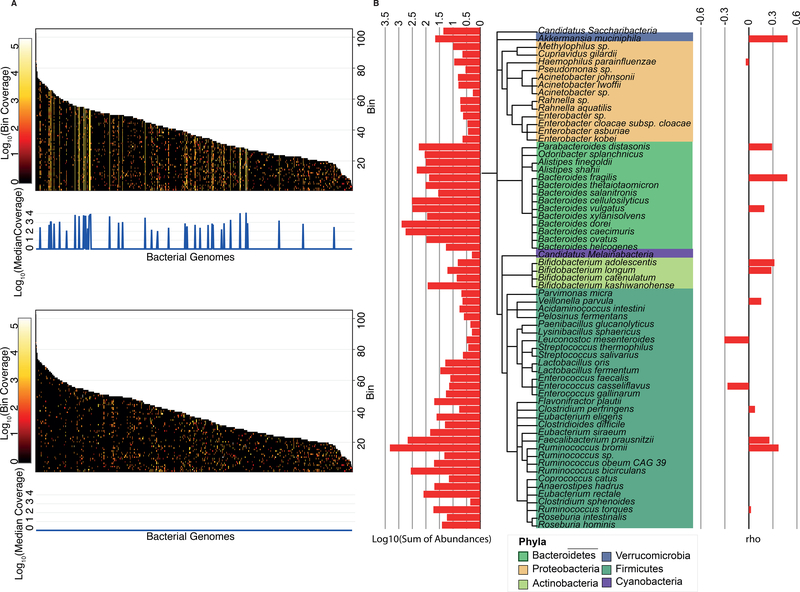
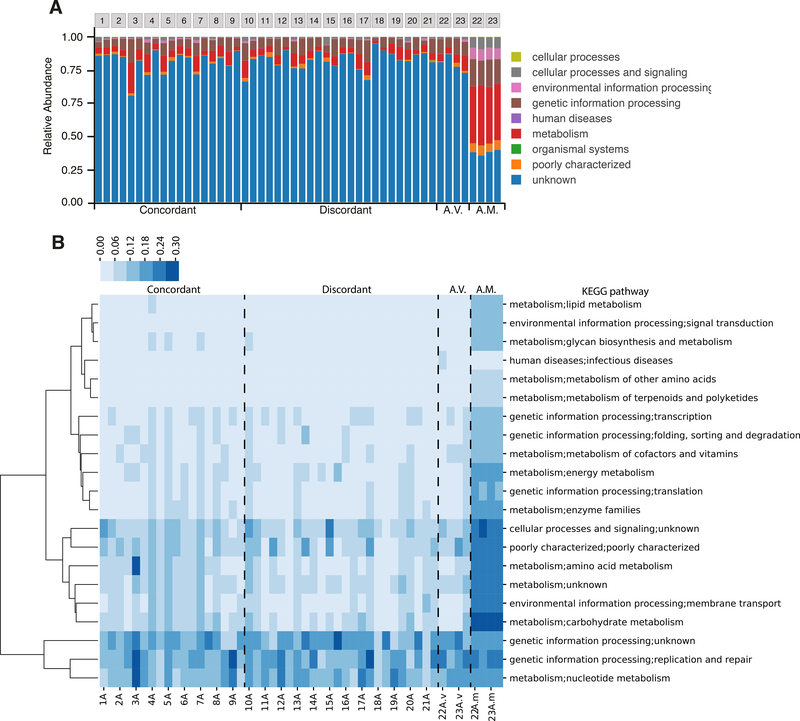
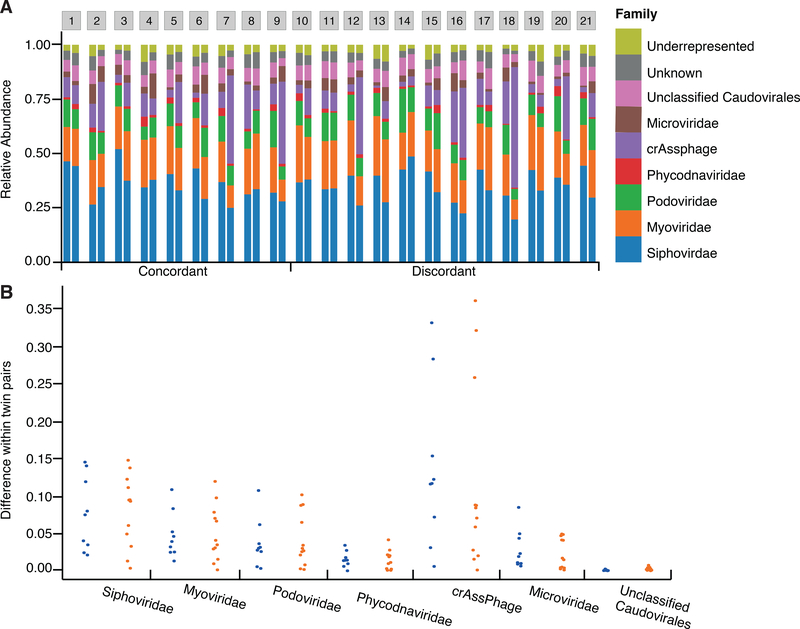
Comment in
-
Gut phages at the centre.Nat Rev Microbiol. 2019 Apr;17(4):195. doi: 10.1038/s41579-019-0174-9. Nat Rev Microbiol. 2019. PMID: 30820034 No abstract available.
References
-
- Alves JMP, de Oliveira AL, Sandberg TOM, Moreno-Gallego JL, de Toledo MAF, de Moura EMM, Oliveira LS, Durham AM, Mehnert DU, Zanotto PM de A, et al. (2016). GenSeed-HMM: A tool for progressive assembly using profile HMMs as seeds and its application in Alpavirinae viral discovery from metagenomic data. Front. Microbiol 7, 269. - PMC - PubMed
-
- Barylski J, Enault F, Dutilh BE, Schuller MBP, Edwards RA, Gillis A, Klumpp J, Knezevic P, Krupovic M, Kuhn JH, et al. (2017). Genomic, proteomic, and phylogenetic analysis of spounaviruses indicates paraphyly of the order Caudovirales. bioRxiv. doi: 10.1101/220434 - DOI
-
- Biller SJ, Schubotz F, Roggensack SE, Thompson AW, Summons RE, and Chisholm SW (2014). Bacterial vesicles in marine ecosystems. Science 343, 183–186. - PubMed
-
- Bray JR, and Curtis JT (1957). An Ordination of the Upland Forest Communities of Southern Wisconsin. Ecol. Monogr 27, 326–349.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
