

Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen
- PMID: 30765700
- PMCID: PMC6376126
- DOI: 10.1038/s41467-019-08734-9
Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen
Abstract
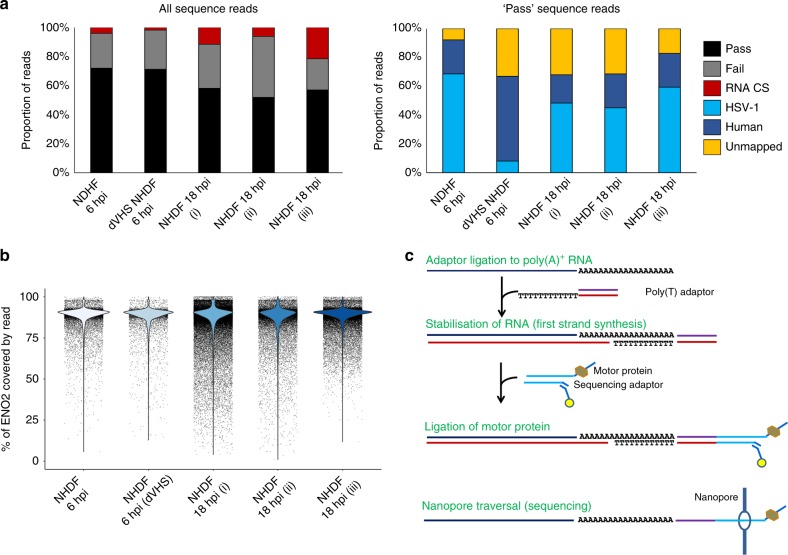
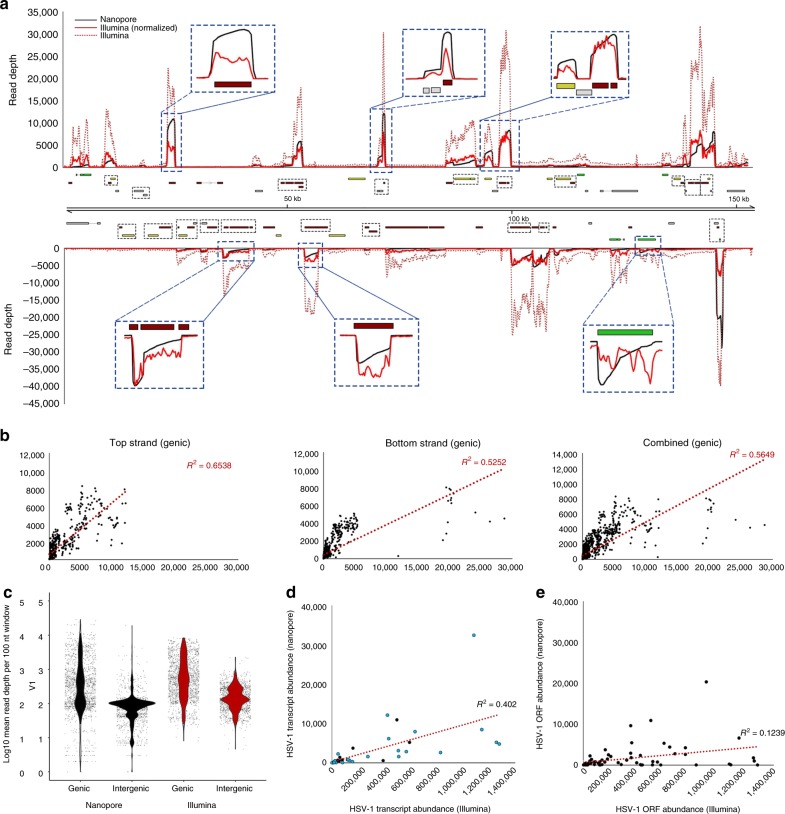
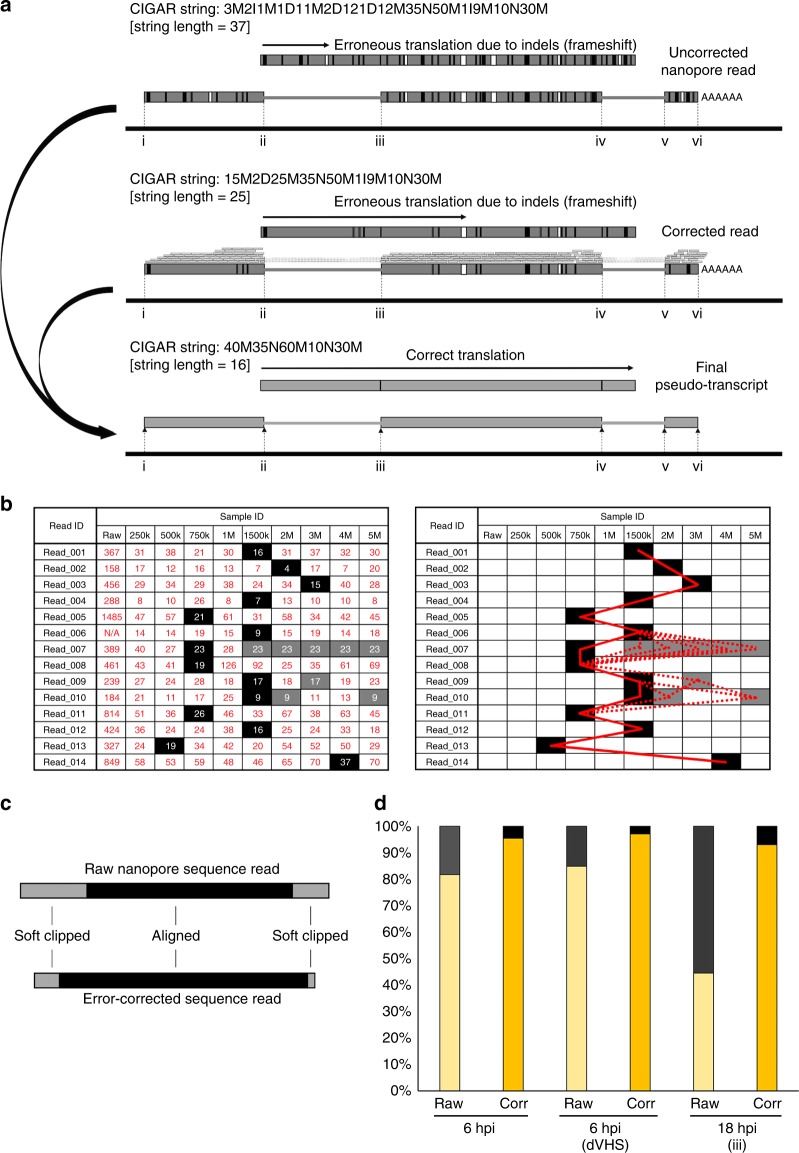
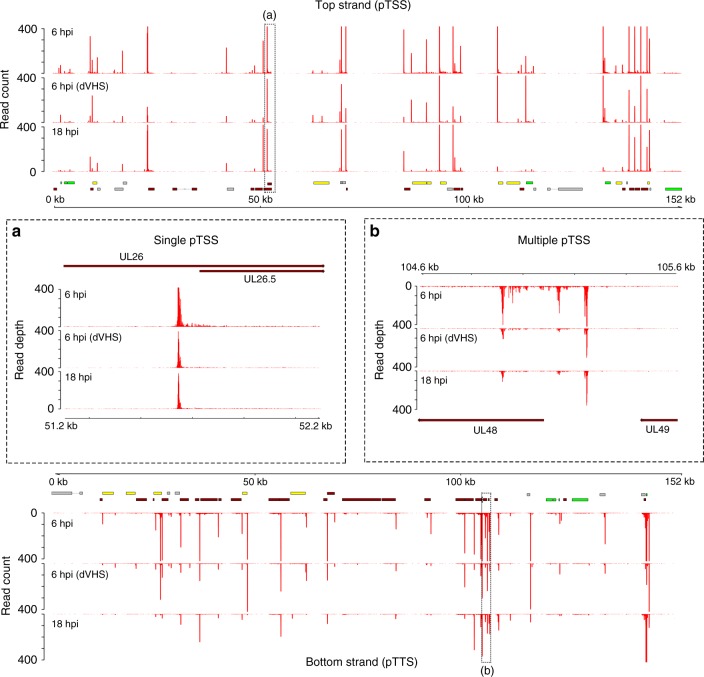
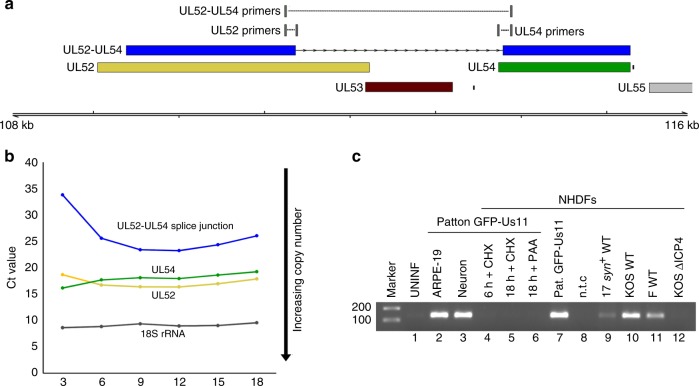
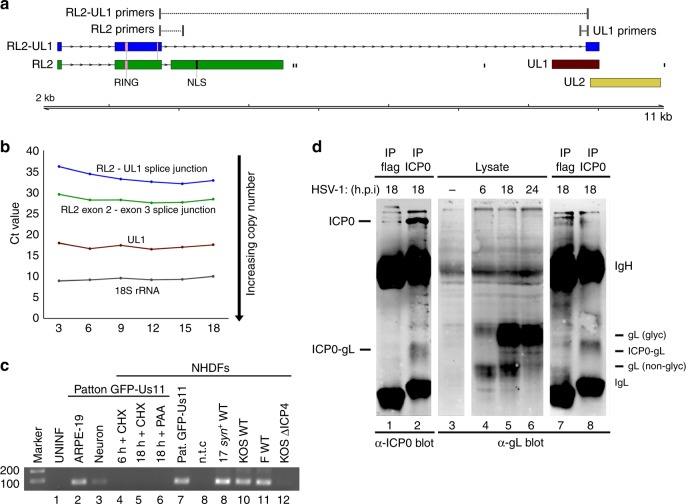
Characterizing complex viral transcriptomes by conventional RNA sequencing approaches is complicated by high gene density, overlapping reading frames, and complex splicing patterns. Direct RNA sequencing (direct RNA-seq) using nanopore arrays offers an exciting alternative whereby individual polyadenylated RNAs are sequenced directly, without the recoding and amplification biases inherent to other sequencing methodologies. Here we use direct RNA-seq to profile the herpes simplex virus type 1 (HSV-1) transcriptome during productive infection of primary cells. We show how direct RNA-seq data can be used to define transcription initiation and RNA cleavage sites associated with all polyadenylated viral RNAs and demonstrate that low level read-through transcription produces a novel class of chimeric HSV-1 transcripts, including a functional mRNA encoding a fusion of the viral E3 ubiquitin ligase ICP0 and viral membrane glycoprotein L. Thus, direct RNA-seq offers a powerful method to characterize the changing transcriptional landscape of viruses with complex genomes.
Conflict of interest statement
The authors declare no competing interests.
Figures

References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
