

A New Panel-Based Next-Generation Sequencing Method for ADME Genes Reveals Novel Associations of Common and Rare Variants With Expression in a Human Liver Cohort
- PMID: 30766545
- PMCID: PMC6365429
- DOI: 10.3389/fgene.2019.00007
A New Panel-Based Next-Generation Sequencing Method for ADME Genes Reveals Novel Associations of Common and Rare Variants With Expression in a Human Liver Cohort
Abstract
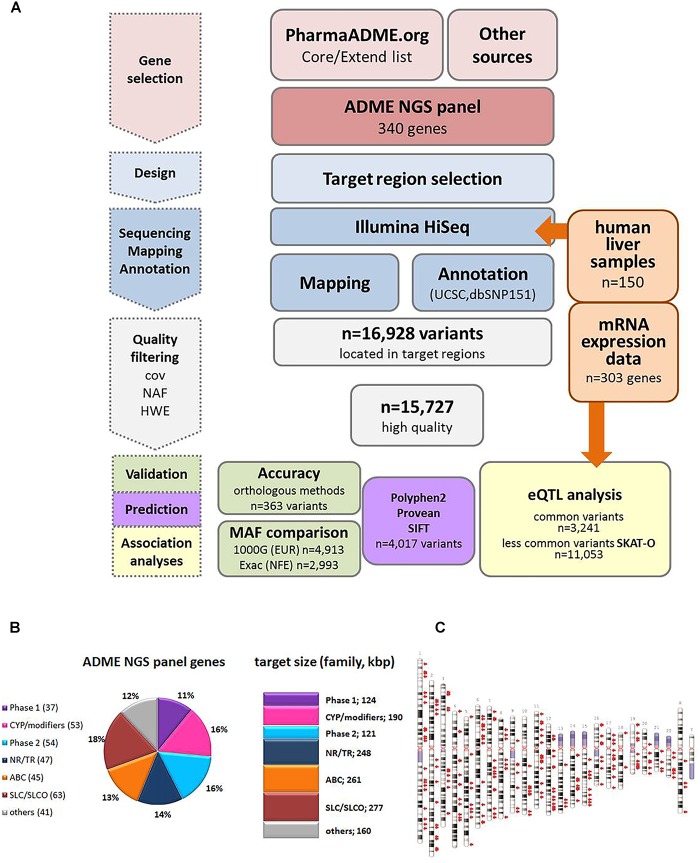
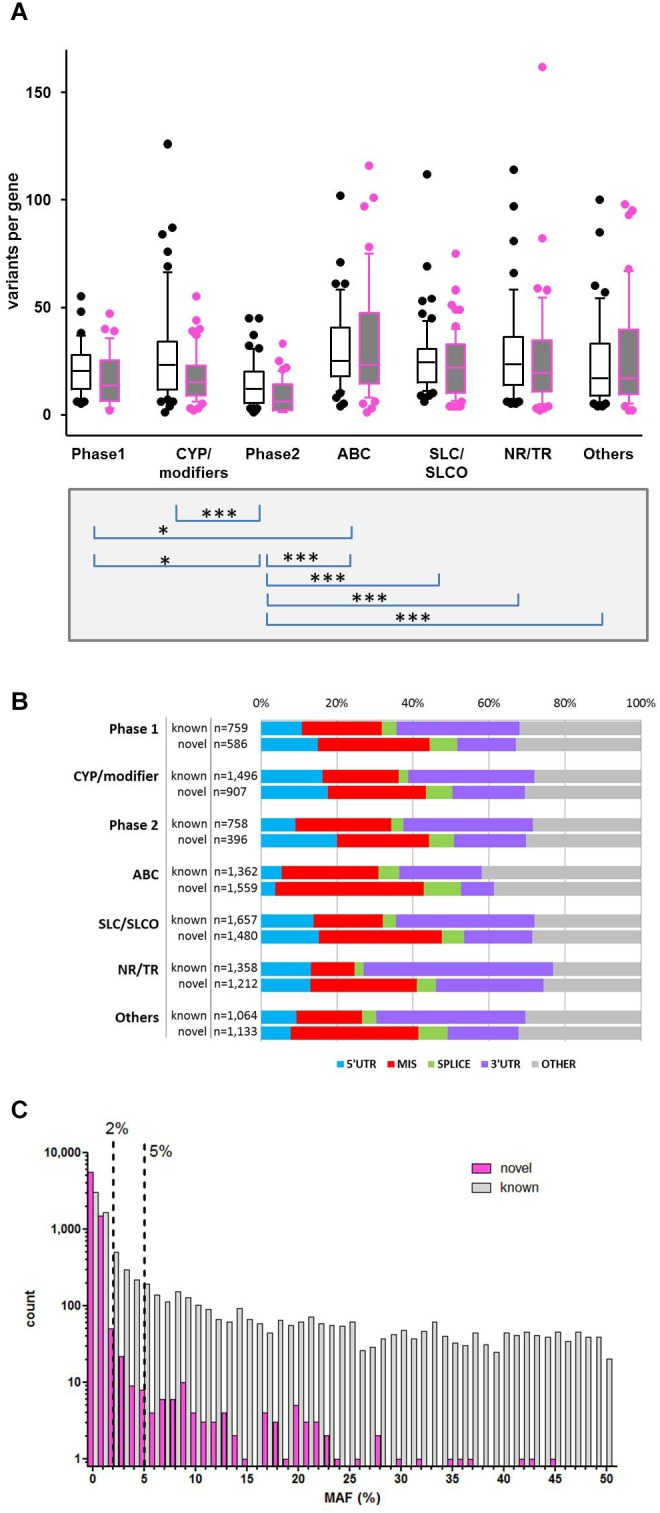
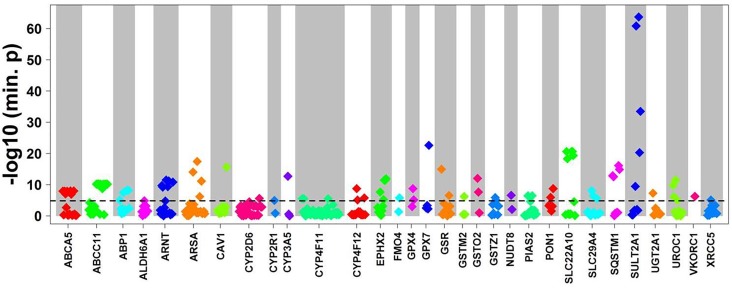
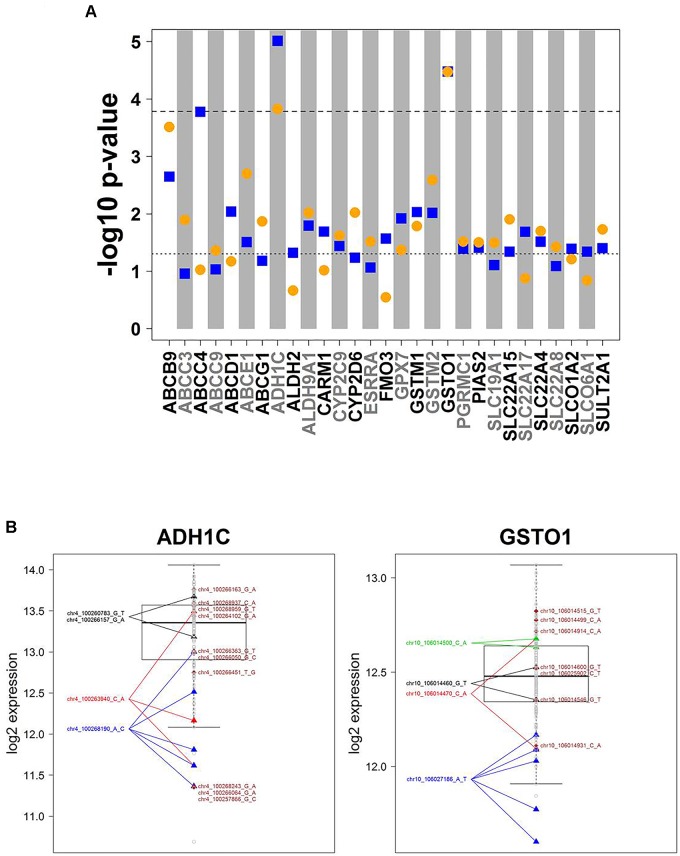
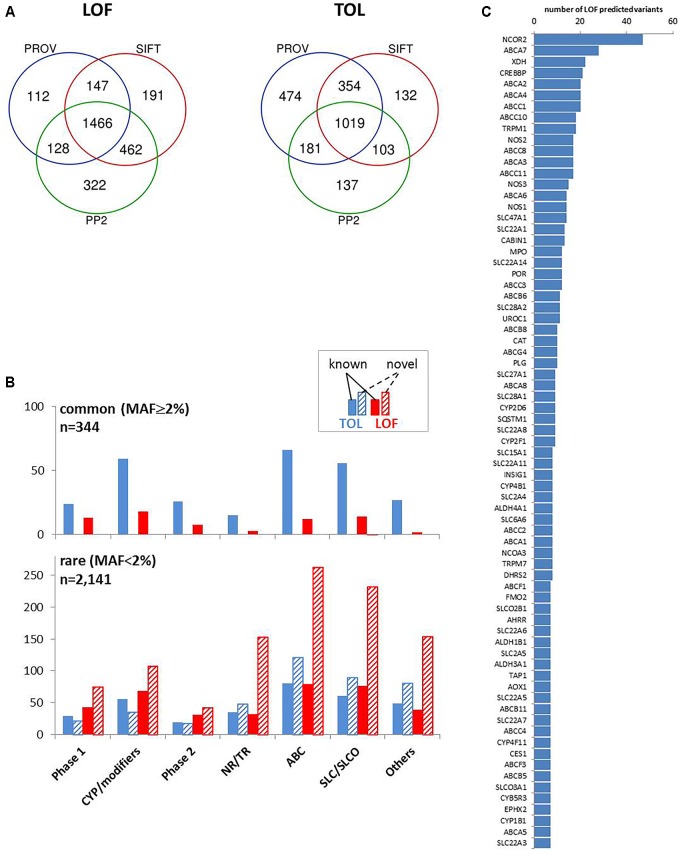
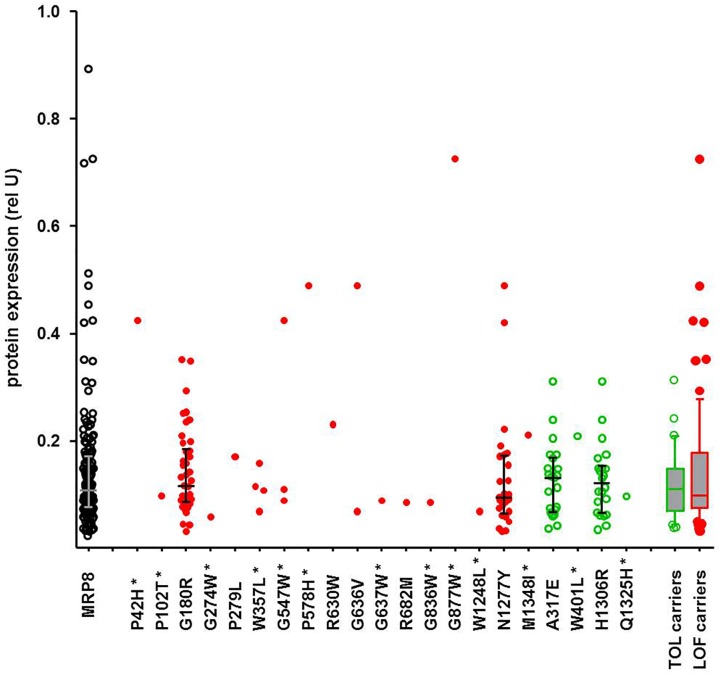
We developed a panel-based NGS pipeline for comprehensive analysis of 340 genes involved in absorption, distribution, metabolism and excretion (ADME) of drugs, other xenobiotics, and endogenous substances. The 340 genes comprised phase I and II enzymes, drug transporters and regulator/modifier genes within their entire coding regions, adjacent intron regions and 5' and 3'UTR regions, resulting in a total panel size of 1,382 kbp. We applied the ADME NGS panel to sequence genomic DNA from 150 Caucasian liver donors with available comprehensive gene expression data. This revealed an average read-depth of 343 (range 27-811), while 99% of the 340 genes were covered on average at least 100-fold. Direct comparison of variant annotation with 363 available genotypes determined independently by other methods revealed an overall accuracy of >99%. Of 15,727 SNV and small INDEL variants, 12,022 had a minor allele frequency (MAF) below 2%, including 8,937 singletons. In total we found 7,273 novel variants. Functional predictions were computed for coding variants (n = 4,017) by three algorithms (Polyphen 2, Provean, and SIFT), resulting in 1,466 variants (36.5%) concordantly predicted to be damaging, while 1,019 variants (25.4%) were predicted to be tolerable. In agreement with other studies we found that less common variants were enriched for deleterious variants. Cis-eQTL analysis of variants with (MAF ≥ 2%) revealed significant associations for 90 variants in 31 genes after Bonferroni correction, most of which were located in non-coding regions. For less common variants (MAF < 2%), we applied the SKAT-O test and identified significant associations to gene expression for ADH1C and GSTO1. Moreover, our data allow comparison of functional predictions with additional phenotypic data to prioritize variants for further analysis.
Keywords: ADME; eQTL analysis; next generation sequencing; pharmacogenomics; rare variants.
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous