

Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria
- PMID: 30767037
- PMCID: PMC6453877
- DOI: 10.1007/s00018-019-03039-y
Exploring the molecular interface between hypoxia-inducible factor signalling and mitochondria
Abstract
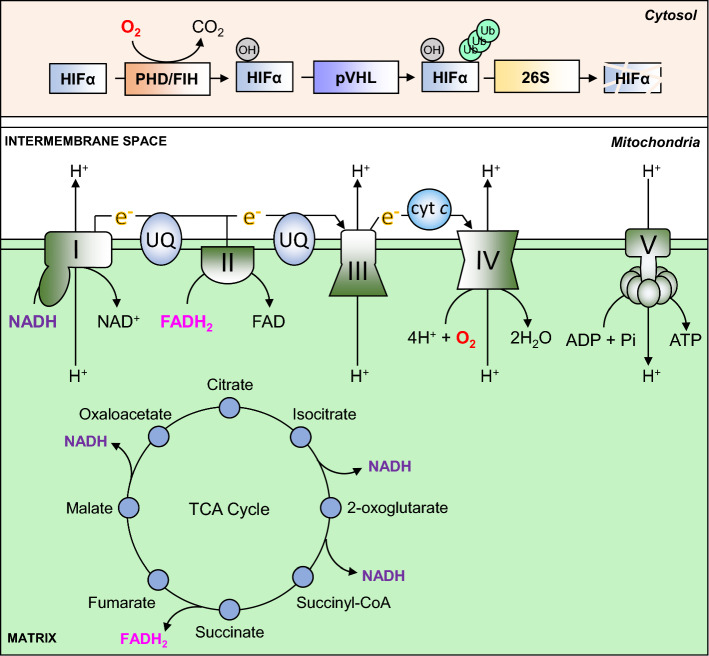
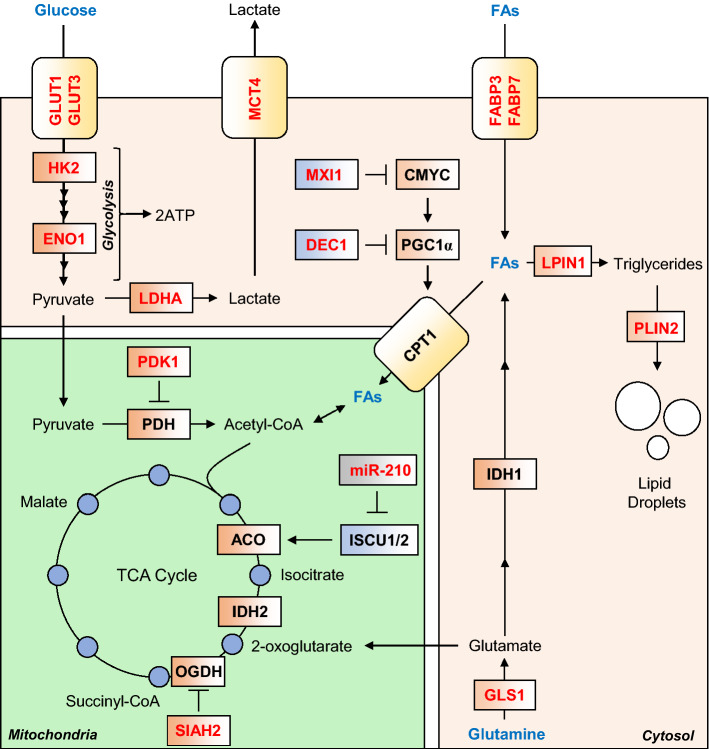
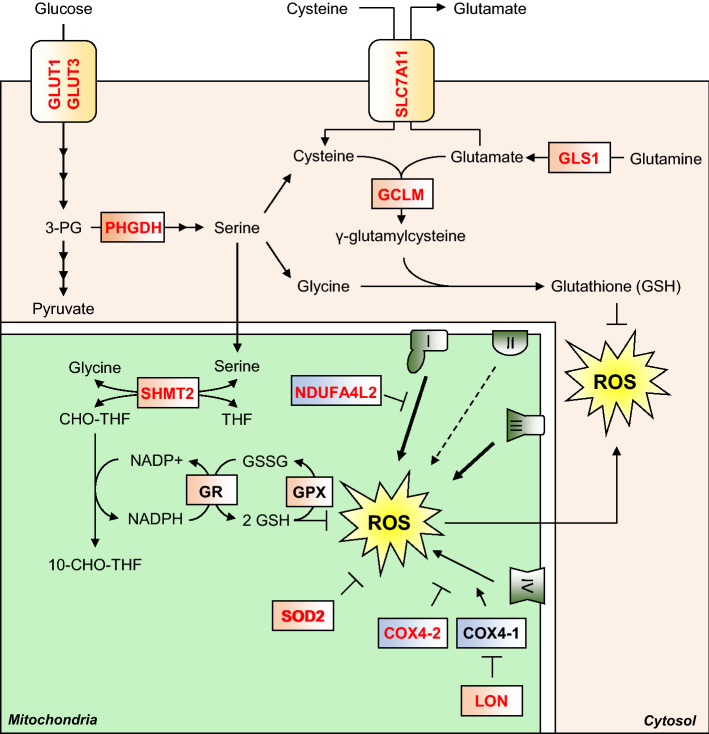
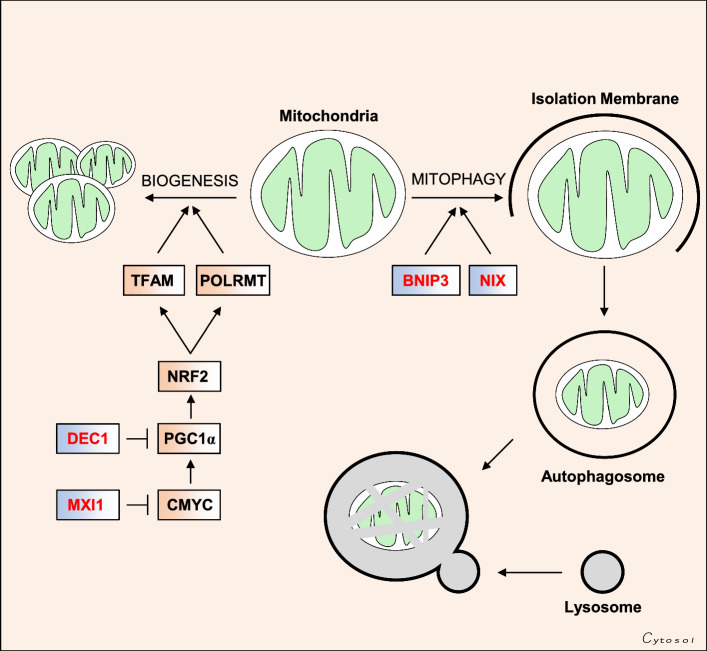
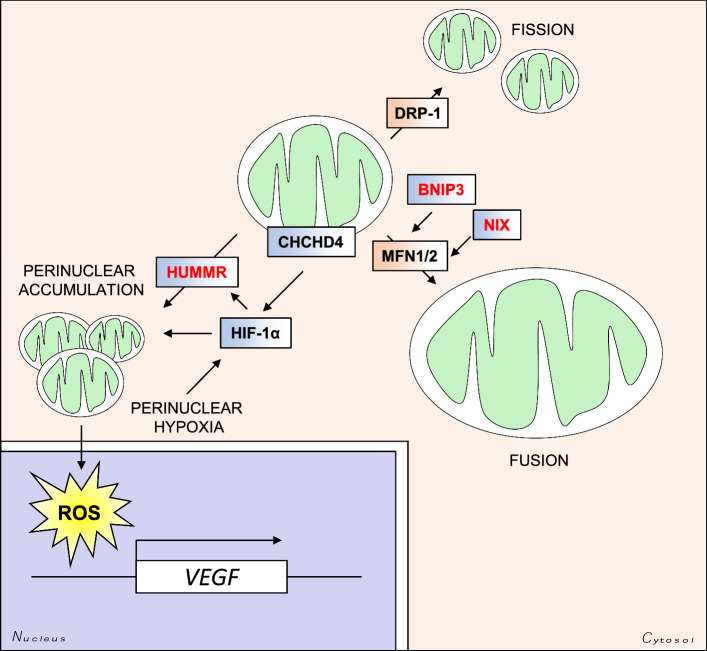
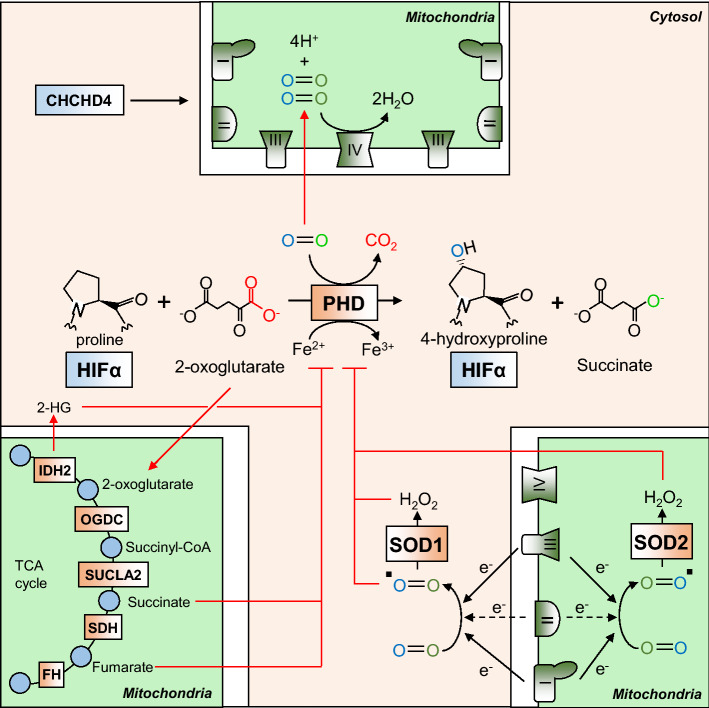
Oxygen is required for the survival of the majority of eukaryotic organisms, as it is important for many cellular processes. Eukaryotic cells utilize oxygen for the production of biochemical energy in the form of adenosine triphosphate (ATP) generated from the catabolism of carbon-rich fuels such as glucose, lipids and glutamine. The intracellular sites of oxygen consumption-coupled ATP production are the mitochondria, double-membraned organelles that provide a dynamic and multifaceted role in cell signalling and metabolism. Highly evolutionarily conserved molecular mechanisms exist to sense and respond to changes in cellular oxygen levels. The primary transcriptional regulators of the response to decreased oxygen levels (hypoxia) are the hypoxia-inducible factors (HIFs), which play important roles in both physiological and pathophysiological contexts. In this review we explore the relationship between HIF-regulated signalling pathways and the mitochondria, including the regulation of mitochondrial metabolism, biogenesis and distribution.
Keywords: HIF; Hypoxia; Metabolism; Mitochondrial biogenesis; Oxphos; Oxygen; Respiratory chain.
Figures
Similar articles
-
Mitochondrial regulation of oxygen sensing.Adv Exp Med Biol. 2010;661:339-54. doi: 10.1007/978-1-60761-500-2_22. Adv Exp Med Biol. 2010. PMID: 20204741 Review.
-
Cellular and molecular mechanisms in the hypoxic tissue: role of HIF-1 and ROS.Cell Biochem Funct. 2013 Aug;31(6):451-9. doi: 10.1002/cbf.2985. Epub 2013 Jun 13. Cell Biochem Funct. 2013. PMID: 23760768 Review.
-
Differential regulation of HIF-mediated pathways increases mitochondrial metabolism and ATP production in hypoxic osteoclasts.J Pathol. 2013 Apr;229(5):755-64. doi: 10.1002/path.4159. J Pathol. 2013. PMID: 23303559 Free PMC article.
-
Mitochondria and cellular oxygen sensing in the HIF pathway.Biochem J. 2008 Jan 1;409(1):19-26. doi: 10.1042/BJ20071249. Biochem J. 2008. PMID: 18062771 Review.
-
Hypoxia-inducible factor-1α activation improves renal oxygenation and mitochondrial function in early chronic kidney disease.Am J Physiol Renal Physiol. 2017 Aug 1;313(2):F282-F290. doi: 10.1152/ajprenal.00579.2016. Epub 2017 Mar 22. Am J Physiol Renal Physiol. 2017. PMID: 28331062 Free PMC article.
Cited by
-
Quercetin relieves compression-induced cell death and lumbar disc degeneration by stabilizing HIF1A protein.Heliyon. 2024 Sep 2;10(17):e37349. doi: 10.1016/j.heliyon.2024.e37349. eCollection 2024 Sep 15. Heliyon. 2024. PMID: 39296087 Free PMC article.
-
Cachexia as Evidence of the Mechanisms of Resistance and Tolerance during the Evolution of Cancer Disease.Int J Mol Sci. 2021 Mar 12;22(6):2890. doi: 10.3390/ijms22062890. Int J Mol Sci. 2021. PMID: 33809200 Free PMC article. Review.
-
Cytoskeletal Arrest: An Anoxia Tolerance Mechanism.Metabolites. 2021 Aug 23;11(8):561. doi: 10.3390/metabo11080561. Metabolites. 2021. PMID: 34436502 Free PMC article. Review.
-
Genome-wide CRISPR/Cas9 deletion screen defines mitochondrial gene essentiality and identifies routes for tumour cell viability in hypoxia.Commun Biol. 2021 May 21;4(1):615. doi: 10.1038/s42003-021-02098-x. Commun Biol. 2021. PMID: 34021238 Free PMC article.
-
Nicotinamide Antagonizes Lipopolysaccharide-Induced Hypoxic Cell Signals in Human Macrophages.J Immunol. 2023 Jul 15;211(2):261-273. doi: 10.4049/jimmunol.2200552. J Immunol. 2023. PMID: 37314413 Free PMC article.
References
-
- Wallimann T, Wyss M, Brdiczka D, Nicolay K, Eppenberger HM. Intracellular compartmentation, structure and function of creatine kinase isoenzymes in tissues with high and fluctuating energy demands: the ‘phosphocreatine circuit’ for cellular energy homeostasis. Biochem J. 1992;281(Pt 1):21–40. doi: 10.1042/bj2810021. - DOI - PMC - PubMed
-
- Carlile MJ, Watkinson SC, Gooday GW. The fungi. 2. San Diego: Academic Press; 2001.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
