

DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks
- PMID: 30768156
- PMCID: PMC6748780
- DOI: 10.1093/bioinformatics/btz111
DeepAffinity: interpretable deep learning of compound-protein affinity through unified recurrent and convolutional neural networks
Abstract
Motivation: Drug discovery demands rapid quantification of compound-protein interaction (CPI). However, there is a lack of methods that can predict compound-protein affinity from sequences alone with high applicability, accuracy and interpretability.
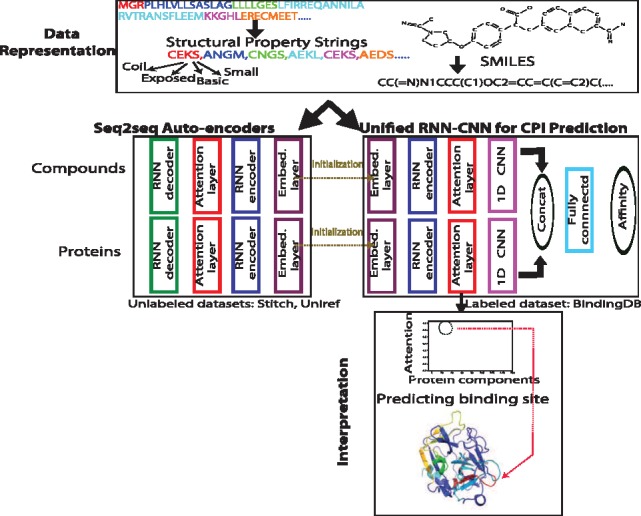
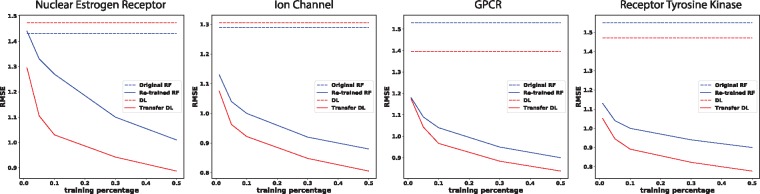
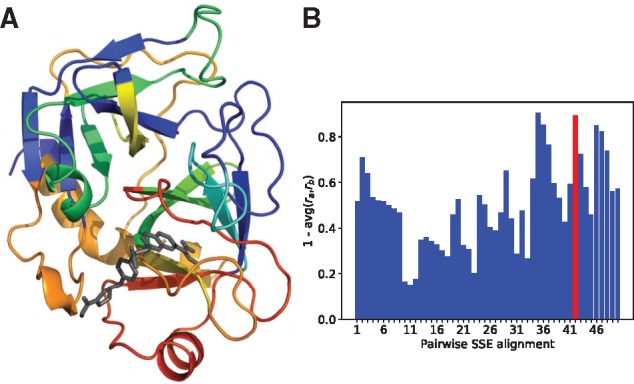
Results: We present a seamless integration of domain knowledges and learning-based approaches. Under novel representations of structurally annotated protein sequences, a semi-supervised deep learning model that unifies recurrent and convolutional neural networks has been proposed to exploit both unlabeled and labeled data, for jointly encoding molecular representations and predicting affinities. Our representations and models outperform conventional options in achieving relative error in IC50 within 5-fold for test cases and 20-fold for protein classes not included for training. Performances for new protein classes with few labeled data are further improved by transfer learning. Furthermore, separate and joint attention mechanisms are developed and embedded to our model to add to its interpretability, as illustrated in case studies for predicting and explaining selective drug-target interactions. Lastly, alternative representations using protein sequences or compound graphs and a unified RNN/GCNN-CNN model using graph CNN (GCNN) are also explored to reveal algorithmic challenges ahead.
Availability and implementation: Data and source codes are available at https://github.com/Shen-Lab/DeepAffinity.
Supplementary information: Supplementary data are available at Bioinformatics online.
© The Author(s) 2019. Published by Oxford University Press. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures
References
-
- Brandstetter H. et al. (1996) X-ray structure of active site-inhibited clotting factor xa implications for drug design and substrate recognition. J. Biol. Chem., 271, 29988–29992. - PubMed
-
- Chen X. et al. (2016) Drug–target interaction prediction: databases, web servers and computational models. Brief. Bioinf., 17, 696–712. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
