

Development of a Sensitive, Scalable Method for Spatial, Cell-Type-Resolved Proteomics of the Human Brain
- PMID: 30768908
- PMCID: PMC6456870
- DOI: 10.1021/acs.jproteome.8b00981
Development of a Sensitive, Scalable Method for Spatial, Cell-Type-Resolved Proteomics of the Human Brain
Abstract
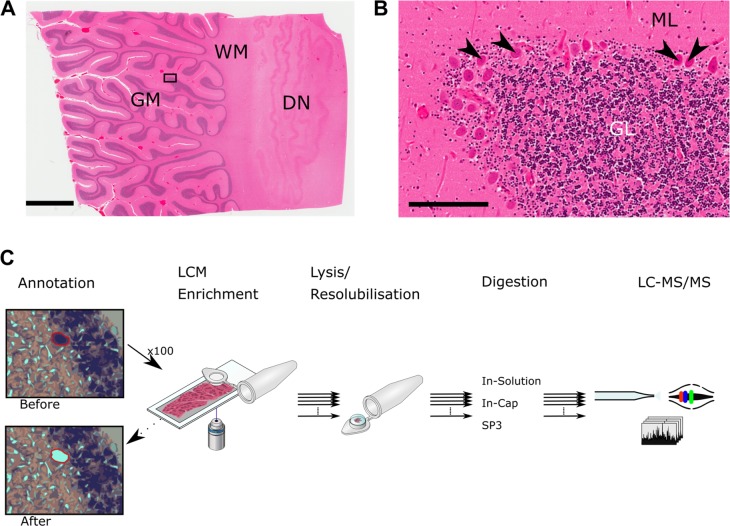
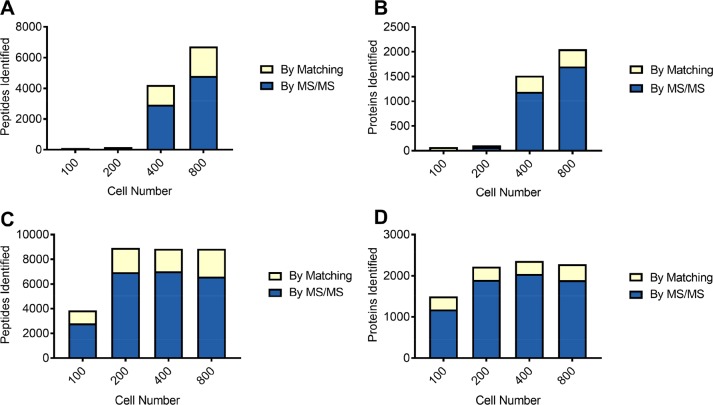
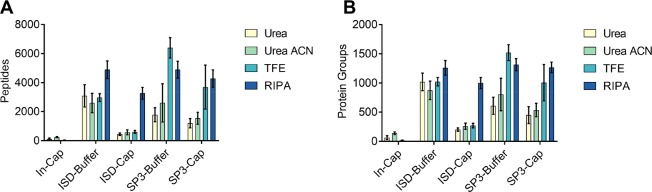
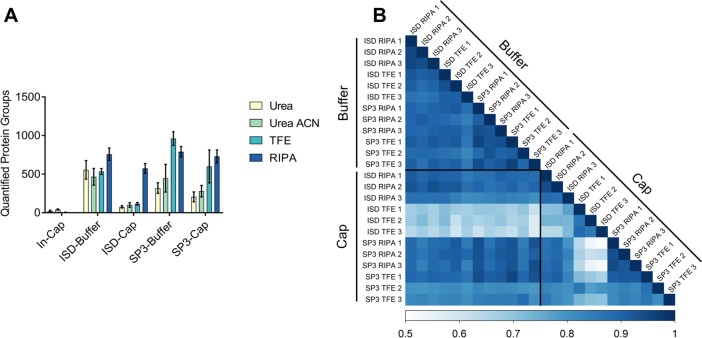
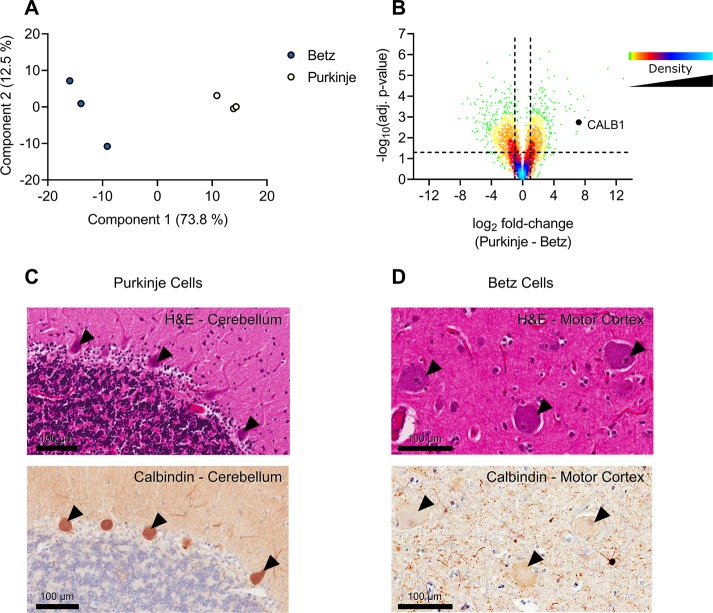
While nearly comprehensive proteome coverage can be achieved from bulk tissue or cultured cells, the data usually lacks spatial resolution. As a result, tissue based proteomics averages protein abundance across multiple cell types and/or localizations. With proteomics platforms lacking sensitivity and throughput to undertake deep single-cell proteome studies in order to resolve spatial or cell type dependent protein expression gradients within tissue, proteome analysis has been combined with sorting techniques to enrich for certain cell populations. However, the spatial resolution and context is lost after cell sorting. Here, we report an optimized method for the proteomic analysis of neurons isolated from post-mortem human brain by laser capture microdissection (LCM). We tested combinations of sample collection methods, lysis buffers and digestion methods to maximize the number of identifications and quantitative performance, identifying 1500 proteins from 60 000 μm2 of 10 μm thick cerebellar molecular layer with excellent reproducibility. To demonstrate the ability of our workflow to resolve cell type specific proteomes within human brain tissue, we isolated sets of individual Betz and Purkinje cells. Both neuronal cell types are involved in motor coordination and were found to express highly specific proteomes to a depth of 2800 to 3600 proteins.
Keywords: LC−MS/MS; brain; laser capture microdissection; spatially resolved proteomics; tissue proteomics.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
