

Systems Biology Analyses Show Hyperactivation of Transforming Growth Factor-β and JNK Signaling Pathways in Esophageal Cancer
- PMID: 30768984
- PMCID: PMC6701681
- DOI: 10.1053/j.gastro.2019.01.263
Systems Biology Analyses Show Hyperactivation of Transforming Growth Factor-β and JNK Signaling Pathways in Esophageal Cancer
Abstract
Background & aims: Esophageal adenocarcinoma (EAC) is resistant to standard chemoradiation treatments, and few targeted therapies are available. We used large-scale tissue profiling and pharmacogenetic analyses to identify deregulated signaling pathways in EAC tissues that might be targeted to slow tumor growth or progression.
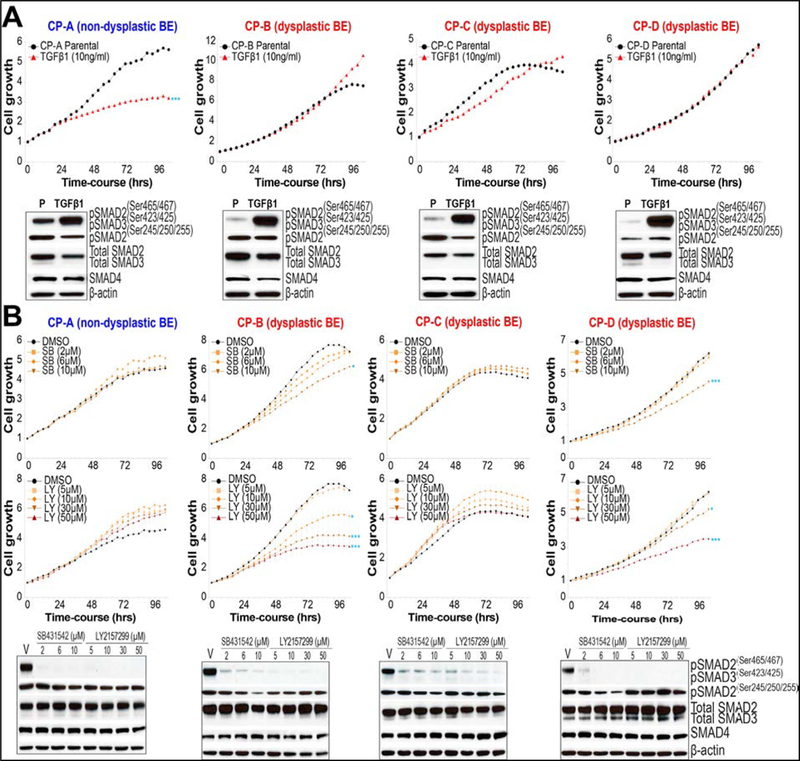
Methods: We collected 397 biopsy specimens from patients with EAC and nonmalignant Barrett's esophagus (BE), with or without dysplasia. We performed RNA-sequencing analyses and used systems biology approaches to identify pathways that are differentially activated in EAC vs nonmalignant dysplastic tissues; pathway activities were confirmed with immunohistochemistry and quantitative real-time polymerase chain reaction analyses of signaling components in patient tissue samples. Human EAC (FLO-1 and EsoAd1), dysplastic BE (CP-B, CP-C, CP-D), and nondysplastic BE (CP-A) cells were incubated with pharmacologic inhibitors or transfected with small interfering RNAs. We measured effects on proliferation, colony formation, migration, and/or growth of xenograft tumors in nude mice.
Results: Comparisons of EAC vs nondysplastic BE tissues showed hyperactivation of transforming growth factor-β (TGFB) and/or Jun N-terminal kinase (JNK) signaling pathways in more than 80% of EAC samples. Immunohistochemical analyses showed increased nuclear localization of phosphorylated JUN and SMAD proteins in EAC tumor tissues compared with nonmalignant tissues. Genes regulated by the TGFB and JNK pathway were overexpressed specifically in EAC and dysplastic BE. Pharmacologic inhibition or knockdown of TGFB or JNK signaling components in EAC cells (FLO-1 or EsoAd1) significantly reduced cell proliferation, colony formation, cell migration, and/or growth of xenograft tumors in mice in a SMAD4-independent manner. Inhibition of the TGFB pathway in BE cell lines reduced the proliferation of dysplastic, but not nondysplastic, cells.
Conclusions: In a transcriptome analysis of EAC and nondysplastic BE tissues, we found the TGFB and JNK signaling pathways to be hyperactivated in EACs and the genes regulated by these pathways to be overexpressed in EAC and dysplastic BE. Inhibiting these pathways in EAC cells reduces their proliferation, migration, and formation of xenograft tumors. Strategies to block the TGFB and JNK signaling pathways might be developed for treatment of EAC.
Keywords: InFlo; LY2157299; PARADIGM; SP600125.
Copyright © 2019 AGA Institute. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
The other authors have no potential conflicts of interest to disclose.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
