

Targeting Alterations in the RAF-MEK Pathway
- PMID: 30770389
- PMCID: PMC6397699
- DOI: 10.1158/2159-8290.CD-18-1321
Targeting Alterations in the RAF-MEK Pathway
Abstract
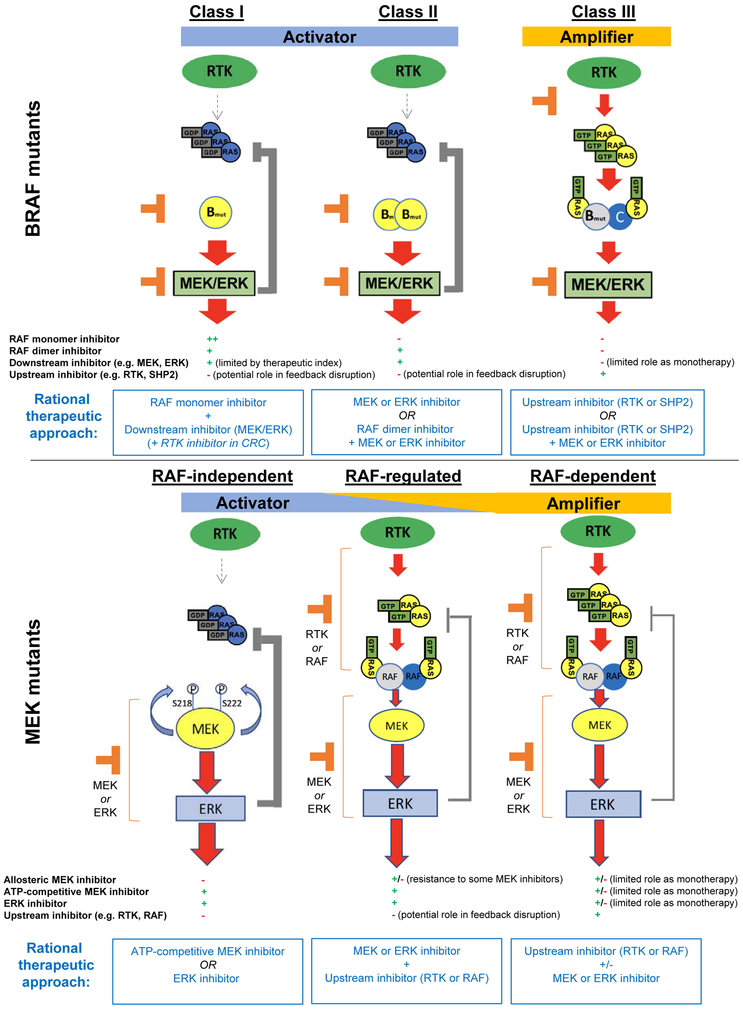
The MAPK pathway is one of the most commonly mutated oncogenic pathways in cancer. Although RAS mutations are the most frequent MAPK alterations, less frequent alterations in downstream components of the pathway, including the RAF and MEK genes, offer promising therapeutic opportunities. In addition to BRAFV600 mutations, for which several approved therapeutic regimens exist, other alterations in the RAF and MEK genes may provide more rare, but tractable, targets. However, recent studies have illustrated the complexity of MAPK signaling and highlighted that distinct alterations in these genes may have strikingly different properties. Understanding the unique functional characteristics of specific RAF and MEK alterations, reviewed herein, will be critical for developing effective therapeutic approaches for these targets. SIGNIFICANCE: Alterations in the RAF and MEK genes represent promising therapeutic targets in multiple cancer types. However, given the unique and complex signaling biology of the MAPK pathway, the diverse array of RAF and MEK alterations observed in cancer can possess distinct functional characteristics. As outlined in this review, understanding the key functional properties of different RAF and MEK alterations is fundamental to selecting the optimal therapeutic approach.
©2019 American Association for Cancer Research.
Conflict of interest statement
Disclosure of Potential Conflicts of Interest:
R.B.C. is a consultant/advisory board member for Amgen, Array Biopharma, Astex Pharmaceuticals, Avidity Biosciences, BMS, Chugai, Fog Pharma, Genentech, LOXO, Merrimack, N-of-one, nRichDx, Roche, Roivant, Shire, Spectrum Pharmaceuticals, Symphogen, Taiho, and Warp Drive Bio; holds equity in Avidity Biosciences and nRichDx; and has received research funding from AstraZeneca and Sanofi. R.Y. has received research funding from Array BioPharma, Genentech, GlaxoSmithKline, and Novartis, and has served as an advisory board member for GlaxoSmithKline.
Figures
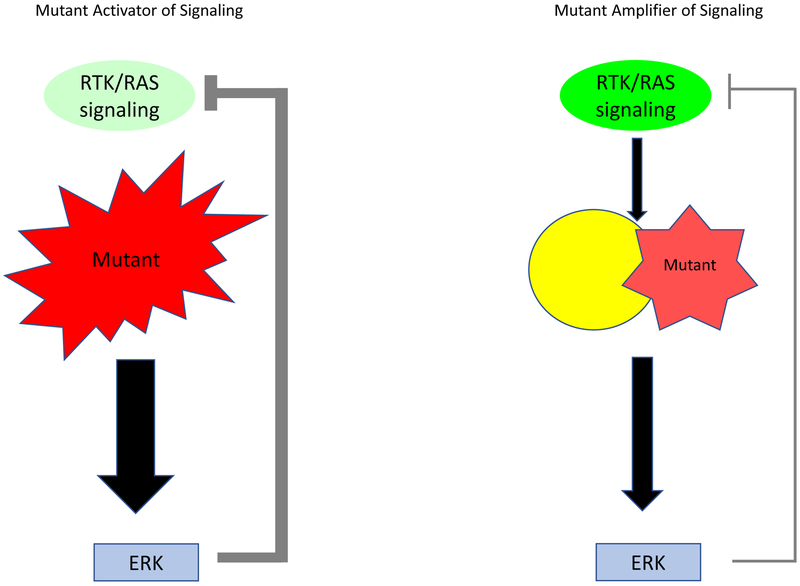
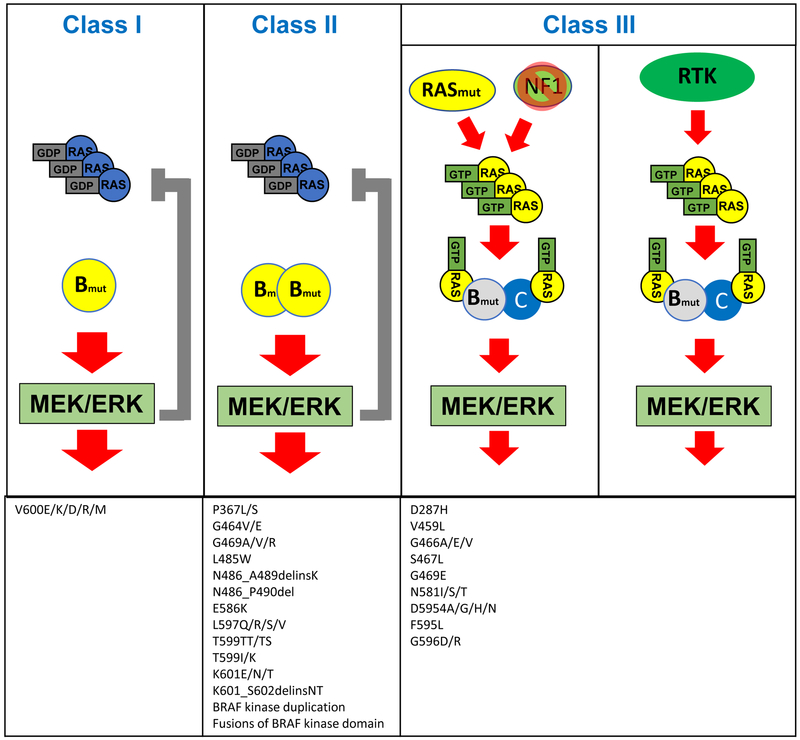
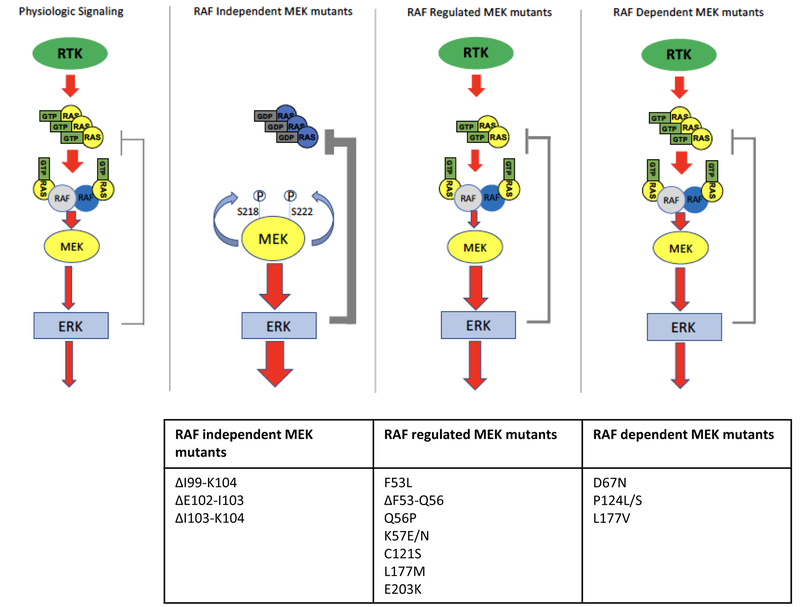
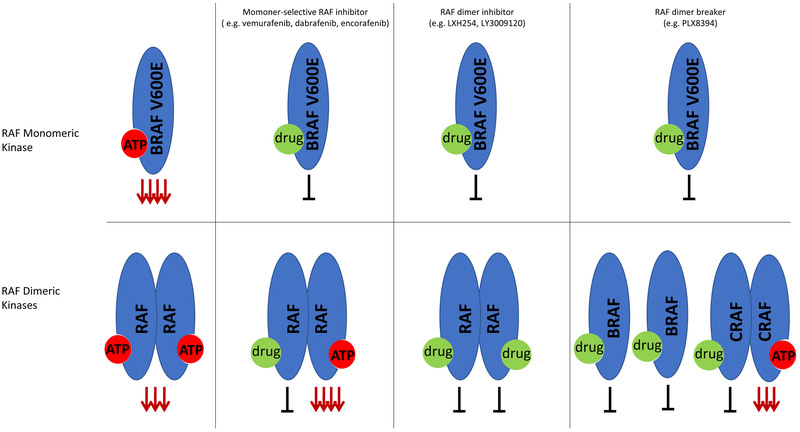
References
-
- Cichowski K, Jacks T. NF1 tumor suppressor gene function: narrowing the GAP. Cell 2001;104(4):593–604. - PubMed
-
- Milburn MV, Tong L, deVos AM, Brunger A, Yamaizumi Z, Nishimura S, et al. Molecular switch for signal transduction: structural differences between active and inactive forms of protooncogenic ras proteins. Science 1990;247(4945):939–45. - PubMed
-
- Scheidig AJ, Burmester C, Goody RS. The pre-hydrolysis state of p21(ras) in complex with GTP: new insights into the role of water molecules in the GTP hydrolysis reaction of ras-like proteins. Structure 1999;7(11):1311–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
