

Smac mimetic suppresses tunicamycin-induced apoptosis via resolution of ER stress
- PMID: 30770792
- PMCID: PMC6377606
- DOI: 10.1038/s41419-019-1381-z
Smac mimetic suppresses tunicamycin-induced apoptosis via resolution of ER stress
Erratum in
-
Correction: Smac mimetic suppresses tunicamycin-induced apoptosis via resolution of ER stress.Cell Death Dis. 2020 Sep 25;11(9):806. doi: 10.1038/s41419-020-02991-z. Cell Death Dis. 2020. PMID: 32978371 Free PMC article.
Abstract
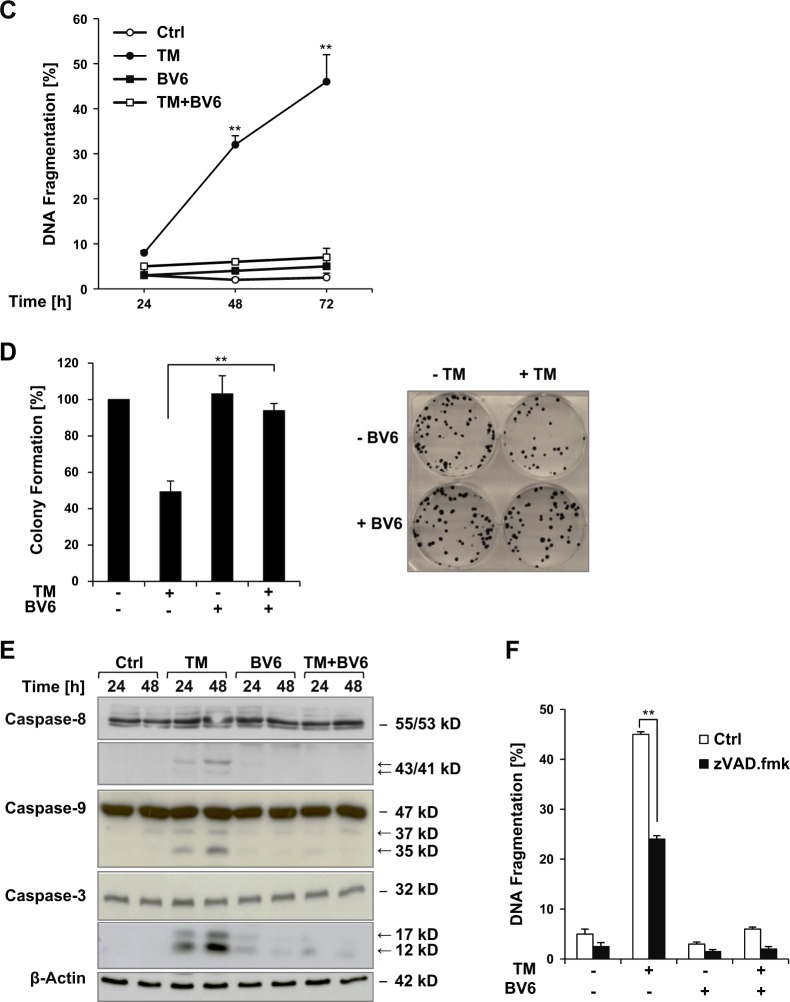
Since Inhibitor of Apoptosis (IAP) proteins have been implicated in cellular adaptation to endoplasmic reticulum (ER) stress, we investigated the regulation of ER stress-induced apoptosis by small-molecule second mitochondria-derived activator of caspase (Smac) mimetics that antagonize IAP proteins. Here, we discover that Smac mimetic suppresses tunicamycin (TM)-induced apoptosis via resolution of the unfolded protein response (UPR) and ER stress. Smac mimetics such as BV6 selectively inhibit apoptosis triggered by pharmacological or genetic inhibition of protein N-glycosylation using TM or knockdown of DPAGT1, the enzyme that catalyzes the first step of protein N-glycosylation. In contrast, BV6 does not rescue cell death induced by other typical ER stressors (i.e., thapsigargin (TG), dithiothreitol, brefeldin A, bortezomib, or 2-deoxyglucose). The protection from TM-triggered apoptosis is found for structurally different Smac mimetics and for genetic knockdown of cellular IAP (cIAP) proteins in several cancer types, underlining the broader relevance. Interestingly, lectin microarray profiling reveals that BV6 counteracts TM-imposed inhibition of protein glycosylation. BV6 consistently abolishes TM-stimulated accumulation of ER stress markers such as glucose-regulated protein 78 (GRP78) and C/EBP homologous protein (CHOP) and reduces protein kinase RNA-like ER kinase (PERK) phosphorylation and X box-binding protein 1 (XBP1) splicing upon TM treatment. BV6-stimulated activation of nuclear factor-κB (NF-κB) contributes to the resolution of ER stress, since NF-κB inhibition by overexpression of dominant-negative IκBα superrepressor counteracts the suppression of TM-stimulated transcriptional activation of CHOP and GRP78 by BV6. Thus, our study is the first to show that Smac mimetic protects from TM-triggered apoptosis by resolving the UPR and ER stress. This provides new insights into the regulation of cellular stress responses by Smac mimetics.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures







References
-
- Lai E, Teodoro T, Volchuk A. Endoplasmic reticulum stress: signaling the unfolded protein response. Physiology (Bethesda). 2007;22:193–201. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
