

Quantification of frequency-dependent genetic architectures in 25 UK Biobank traits reveals action of negative selection
- PMID: 30770844
- PMCID: PMC6377669
- DOI: 10.1038/s41467-019-08424-6
Quantification of frequency-dependent genetic architectures in 25 UK Biobank traits reveals action of negative selection
Abstract
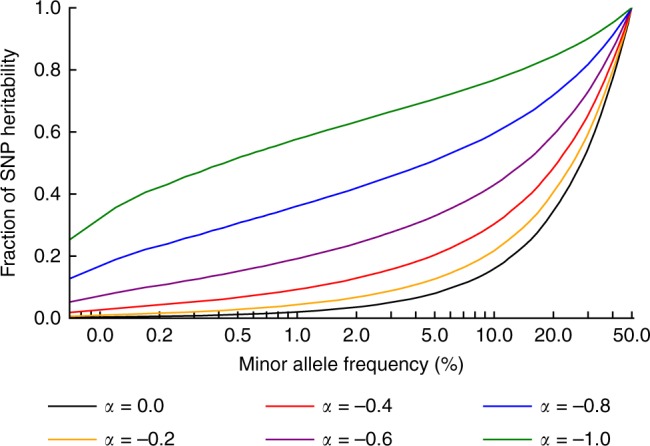
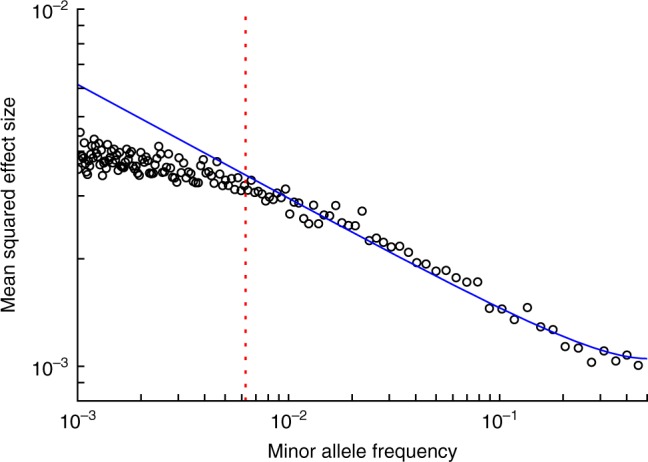
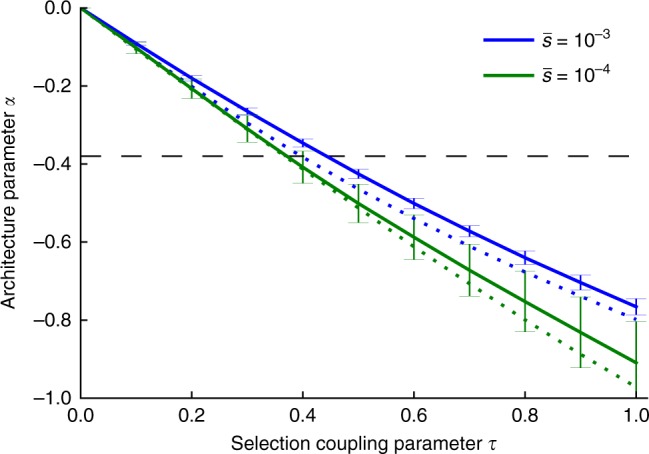
Understanding the role of rare variants is important in elucidating the genetic basis of human disease. Negative selection can cause rare variants to have larger per-allele effect sizes than common variants. Here, we develop a method to estimate the minor allele frequency (MAF) dependence of SNP effect sizes. We use a model in which per-allele effect sizes have variance proportional to [p(1 - p)]α, where p is the MAF and negative values of α imply larger effect sizes for rare variants. We estimate α for 25 UK Biobank diseases and complex traits. All traits produce negative α estimates, with best-fit mean of -0.38 (s.e. 0.02) across traits. Despite larger rare variant effect sizes, rare variants (MAF < 1%) explain less than 10% of total SNP-heritability for most traits analyzed. Using evolutionary modeling and forward simulations, we validate the α model of MAF-dependent trait effects and assess plausible values of relevant evolutionary parameters.
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
