

The Search for Disease-Modifying Therapies in Pulmonary Hypertension
- PMID: 30773044
- PMCID: PMC6714051
- DOI: 10.1177/1074248419829172
The Search for Disease-Modifying Therapies in Pulmonary Hypertension
Abstract
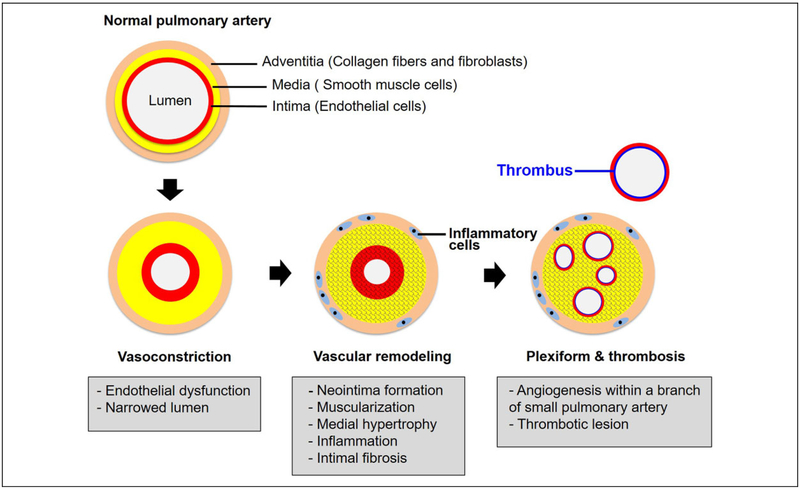
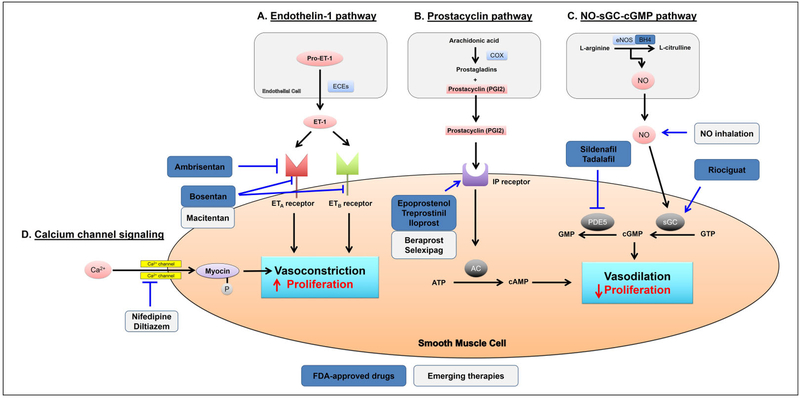
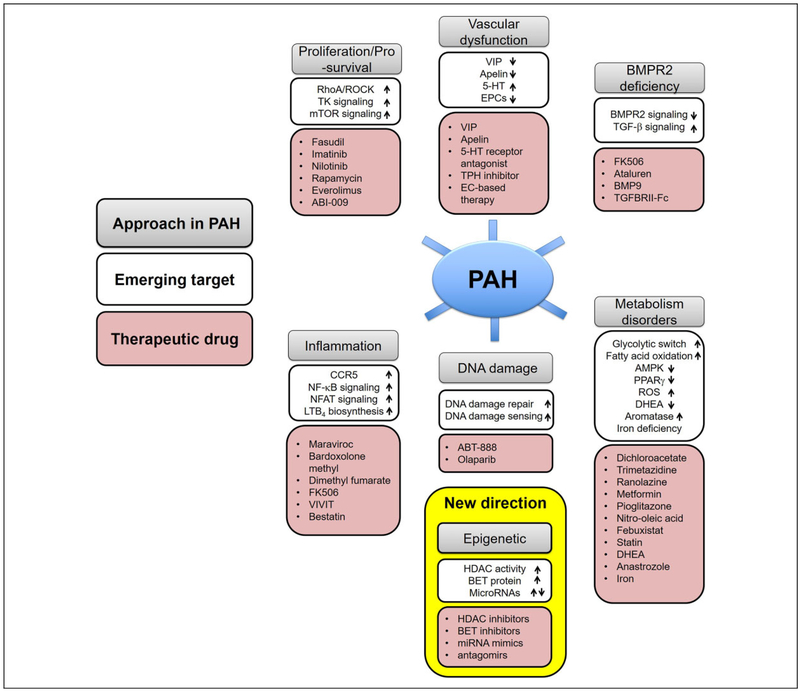
Pulmonary hypertension (PH) and its severe subtype pulmonary arterial hypertension (PAH) encompass a set of multifactorial diseases defined by sustained elevation of pulmonary arterial pressure and pulmonary vascular resistance leading to right ventricular failure and subsequent death. Pulmonary hypertension is characterized by vascular remodeling in association with smooth muscle cell proliferation of the arterioles, medial thickening, and plexiform lesion formation. Despite our recent advances in understanding its pathogenesis and related therapeutic discoveries, PH still remains a progressive disease without a cure. Nevertheless, development of drugs that specifically target molecular pathways involved in disease pathogenesis has led to improvement in life quality and clinical outcomes in patients with PAH. There are presently more than 12 Food and Drug Administration-approved vasodilator drugs in the United States for the treatment of PAH; however, mortality with contemporary therapies remains high. More recently, there have been exuberant efforts to develop new pharmacologic therapies that target the fundamental origins of PH and thus could represent disease-modifying opportunities. This review aims to summarize recent developments on key signaling pathways and molecular targets that drive PH disease progression, with emphasis on new therapeutic options under development.
Keywords: DNA damage; epigenetics; inflammation; metabolism; molecular pathology; proliferation; pulmonary artery hypertension; vascular function.
Conflict of interest statement
Declaration of Conflicting Interests
The author(s) declared the following potential conflicts of interest with respect to the research, authorship, and/or publication of this article. S.Y.C. has served as a consultant for Actelion (Significant), Gilead, Aerpio, Pfizer, and Vivus (Modest). Patent applications (S.Y.C.) have been filed regarding targeting metabolism in pulmonary hypertension.
Figures
References
-
- Hoeper MM, Humbert M, Souza R, et al. A global view of pulmonary hypertension. Lancet Respir Med. 2016;4(4):306–322. - PubMed
-
- Humbert M, Morrell NW, Archer SL, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;430(suppl 12):S13–S24. - PubMed
-
- Tuder RM, Lee SD, Cool CC. Histopathology of pulmonary hypertension. Chest. 1998;114(1):1S–6S. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
